도나-바로우 증후군
Donnai–Barrow syndrome| 도나-바로우 증후군 | |
|---|---|
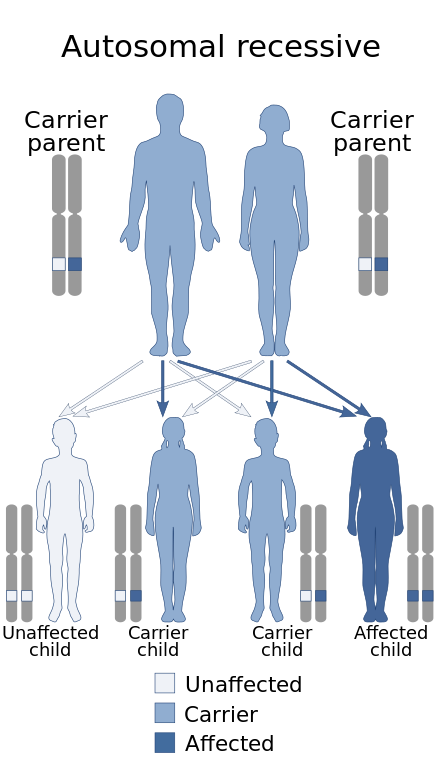 | |
| 도나-바로우 증후군은 상염색체 열성으로 유전됩니다. | |
| 전문 | 의학유전학 |
도나-배로우 증후군은 1993년 Dian Donnai와 Margaret Barrow에 의해 처음 기술된 유전 질환입니다.[1] LRP2와 관련이 있습니다.[2] 그것은 신체의 많은 부분에 영향을 미치는 유전적인 (유전적인) 장애입니다.
발표
이 질환은 아래쪽을 가리키는 바깥쪽 모서리가 있는 눈이 두드러지고 넓게 설정된 눈, 납작한 콧대가 있는 짧은 구근형 코, 뒤로 회전하는 귀, 그리고 과부의 절정의 헤어라인을 포함한 특이한 얼굴 특징을 가지고 있습니다.[3]
도나-바로우 증후군 환자는 내이의 이상으로 인한 심한 난청(신경성 난청)을 가지고 있습니다. 그 외에도 극심한 근시(고도근시), 눈 뒷부분(망막)의 빛에 민감한 조직의 박리나 변질, 진행성 시력 저하 등 시력 문제를 겪는 경우가 많습니다. 일부는 눈의 색이 있는 부분(홍채색종)에 틈이 생기거나 갈라집니다.[3][4]
도나-바로우 증후군이 있는 거의 모든 사람들은 좌뇌와 우뇌를 연결하는 조직(corpus callosum)이 발달되지 않았거나 없습니다. 영향을 받는 사람들은 뇌의 다른 구조적 이상을 가지고 있을 수도 있습니다. 그들은 일반적으로 경도에서 중등도의 지적 장애와 발달 지연을 가지고 있습니다.[3]
도나-배로우 증후군을 가진 사람들은 또한 횡격막 탈장이라고 불리는 흉강(격막)으로부터 복부를 분리하는 근육에 구멍이 있을 수 있습니다. 이 잠재적으로 심각한 선천적 결함은 위와 장이 가슴으로 이동하고 아마도 발달 중인 심장과 폐를 무리지어 다닐 수 있게 합니다. 복부 벽에 있는 복부 기관이 배꼽을 통해 돌출되도록 하는 구멍(임파구)도 영향을 받은 사람에게서 발생할 수 있습니다. 가끔 도나-바로우 증후군을 가진 사람들은 장, 심장 또는 다른 장기의 이상과 척추측만증이 있습니다.[3][4]
원인들
LRP2 유전자의 돌연변이는 도나이-바로우 증후군을 유발합니다. LRP2 유전자는 수용체 기능을 하는 메갈린이라는 단백질을 만드는 지침을 제공합니다. 수용체 단백질은 리간드라고 불리는 특정한 다른 단백질이 열쇠처럼 자물쇠에 들어가는 특정한 부위를 가지고 있습니다. 리간드와 그 수용체는 함께 세포 발달과 기능에 영향을 미치는 신호를 유발합니다. 메갈린은 비타민 A와 D의 흡수, 면역 기능, 스트레스 반응, 혈류 속 지방의 운반 등 다양한 신체 과정에 관여하는 많은 리간드를 가지고 있습니다.[3][4]
메갈린은 신체의 표면과 공동(상피세포)을 따라 늘어선 세포막에 박혀 있습니다. 수용체는 리간드가 세포 표면에서 세포 안으로 이동하는 것을 돕습니다. 뇌와 척수(중추신경계), 눈, 귀, 폐, 장, 생식기, 소변이 만들어지는 신장의 작은 관(신세뇨관) 등 신체의 많은 부분의 발달과 기능에 적극적입니다.[3]
도나-바로우 증후군을 일으키는 LRP2 유전자 돌연변이는 기능성 메갈린 단백질이 없는 것으로 추정됩니다. 신장 세뇨관에 기능성 메갈린이 부족하면 메갈린의 다양한 리간드가 혈류로 다시 흡수되지 않고 소변으로 배출됩니다. 도나-배로우 증후군의 특징은 아마도 메갈린이 이러한 리간드를 흡수하는 것을 도울 수 없거나 생화학적 신호 경로의 파괴 또는 비기능적인 메갈린 단백질의 다른 영향에 의해 발생합니다. 그러나 이러한 이상이 어떻게 장애의 구체적인 징후와 증상을 초래하는지는 불분명합니다.[3]
이전에 별도의 질환으로 분류되었던 FOAR(facio-culo-acoustico-renal) 증후군도 LRP2 돌연변이에 의해 발생하는 것으로 밝혀졌습니다. FOAR 증후군은 이제 도나-바로우 증후군과 같은 장애로 여겨집니다.[3][4]
유전변이
이 상태는 상염색체 열성 패턴으로 유전되는데, 이것은 각 세포에 있는 유전자의 두 사본이 모두 돌연변이를 가지고 있다는 것을 의미합니다. 거의 모든 경우에, 상염색체 열성 질환을 가진 사람의 부모는 각각 돌연변이 유전자의 사본을 하나씩 가지고 있지만, 일반적으로 그 질환의 징후와 증상을 보이지 않습니다.[3][4]
도나-바로우 증후군을 앓고 있는 한 사람은 UPD(Uniparent Disomy)라고 불리는 유전적 변화의 결과로 그의 아버지로부터 돌연변이 유전자의 두 사본을 모두 물려받은 것으로 밝혀졌습니다. UPD는 어떤 사람이 한 부모로부터 염색체 또는 염색체의 일부의 사본을 두 개 받고 다른 부모로부터는 사본을 받지 않을 때 발생합니다. UPD는 난자나 정자 세포가 형성되는 동안 무작위적인 사건으로 발생하거나 초기 태아 발달에서 발생할 수 있습니다.[3][4]
진단.
관리
역학
도나-바로우 증후군은 희귀한 장애인 것으로 보입니다. 세계 여러 지역에서 수십 명의 감염자가 보고되었습니다.[3][4]
참고문헌
- ^ Donnai, D; Barrow, M (1993). "Diaphragmatic hernia, exomphalos, absent corpus callosum, hypertelorism, myopia, and sensorineural deafness: a newly recognized autosomal recessive disorder?". American Journal of Medical Genetics. 47 (5): 679–682. doi:10.1002/ajmg.1320470518. PMID 8266995.
- ^ Kantarci S; Al-Gazali L; Hill RS; et al. (August 2007). "Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai-Barrow and facio-oculo-acoustico-renal syndromes". Nat. Genet. 39 (8): 957–9. doi:10.1038/ng2063. PMC 2891728. PMID 17632512.
- ^ a b c d e f g h i j k "Donnai-Barrow syndrome" (online). MedlinePlus. U.S. National Library of Medicine of the National Institutes of Health. 1 April 2013. Archived from the original on 2020-10-21. Retrieved 8 May 2021. 이 기사는 공공 영역에 있는 미국 국립 의학 도서관의 텍스트를 통합합니다.
- ^ a b c d e f g 아래 자료들은 도나-바로우 증후군에 대한 이 기사의 상태 요약을 개발하는 데 사용되었습니다.
- Chassaing, N; Lacombe, D; Carles, D; Calvas, P; Saura, R; Bieth, E (2003). "Donnai-Barrow syndrome: four additional patients". American Journal of Medical Genetics Part A. 121A (3): 258–62. doi:10.1002/ajmg.a.20266. PMID 12923867. S2CID 38409652.
- Chen, CP (2007). "Syndromes and disorders associated with omphalocele (III): single gene disorders, neural tube defects, diaphragmatic defects and others". Taiwanese Journal of Obstetrics & Gynecology. 46 (2): 111–20. doi:10.1016/S1028-4559(07)60004-7. PMID 17638618.
- Kantarci, S; Donnai, D; Noonan, KM; Pober, BR; Pagon, RA; Bird, TC; Dolan, CR; Stephens, K (1993). "Donnai-Barrow Syndrome". PMID 20301732.
{{cite journal}}: 저널 인용 요구사항journal=(도와주세요) - Kantarci, S; Al-Gazali, L; Hill, RS; Donnai, D; Black, GC; Bieth, E; Chassaing, N; Lacombe, D; Devriendt, K (2007). "Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai-Barrow and facio-oculo-acoustico-renal syndromes". Nature Genetics. 39 (8): 957–9. doi:10.1038/ng2063. PMC 2891728. PMID 17632512.
- Kantarci, S; Ragge, NK; Thomas, NS; Robinson, DO; Noonan, KM; Russell, MK; Donnai, D; Raymond, FL; Walsh, CA (2008). "Donnai-Barrow syndrome (DBS/FOAR) in a child with a homozygous LRP2 mutation due to complete chromosome 2 paternal isodisomy". American Journal of Medical Genetics Part A. 146A (14): 1842–7. doi:10.1002/ajmg.a.32381. PMC 2891749. PMID 18553518.
- Patel, N; Hejkal, T; Katz, A; Margalit, E (2007). "Ocular manifestations of Donnai-Barrow syndrome". Journal of Child Neurology. 22 (4): 462–4. doi:10.1177/0883073807301933. PMID 17621530. S2CID 28446743.
- Pober, BR; Longoni, M; Noonan, KM (2009). "A review of Donnai-Barrow and facio-oculo-acoustico-renal (DB/FOAR) syndrome: clinical features and differential diagnosis". Birth Defects Research. Part A, Clinical and Molecular Teratology. 85 (1): 76–81. doi:10.1002/bdra.20534. PMC 2882234. PMID 19089858.
- "Bulleted reference list (above)" (online). U.S. National Library of Medicine of the National Institutes of Health. May 2009. Retrieved 2010-12-19.
- 이 문서는 공용 도메인에 있는 미국 국립 의학 도서관(NLM 페이지 URL)의 텍스트를 통합합니다.