Chimie minérale

| Partie de | |
|---|---|
| Pratiqué par |
Chimiste inorganicien (d) |
| Objet |
La chimie minérale, aussi appelée chimie inorganique (par traduction littérale de l'anglais), est la branche la plus ancienne de la chimie[1]. Elle comporte l'étude des divers corps simples existant dans la nature ou obtenus artificiellement et celle des composés qu'ils engendrent en réagissant les uns sur les autres, à l'exception des combinaisons avec le carbone qui sont étudiées à part et font l'objet de la chimie organique[2]. Cependant quelques composés simples du carbone (certains oxydes de carbone[3], les carbonates, bicarbonates et cyanures ioniques, les carbures, excepté les hydrocarbures)[4] sont classés parmi les composés inorganiques. Une étude particulière est celle des propriétés et de la synthèse des composés inorganiques artificiels[5], laquelle inclut les composés organométalliques[6],[7]. Ce domaine couvre tous les composés chimiques à l'exception des myriades de composés organiques qui sont basés sur un squelette carboné et comportent habituellement des liaisons C-H. À l'origine basée sur des arguments historiques, cette distinction est de nos jours loin d'être absolue, et de nombreux recouvrements existent en particulier dans le domaine de la chimie organométallique. La chimie inorganique est un domaine de recherche actif actuellement et possède des applications dans la plupart des aspects de l'industrie chimique, en particulier en catalyse, science des matériaux, pigments, surfactants, chimie médicinale, carburants, chimie de l'environnement et agriculture[8].
Histoire
[modifier | modifier le code]Bien avant que l'alchimie ne devienne un sujet d'étude, de nombreuses réactions chimiques employant des composés inorganiques étaient utilisées et leurs produits utilisés dans la vie de tous les jours. L'âge du bronze et l'âge du fer correspondent à des périodes de la Protohistoire au cours desquelles la métallurgie du bronze (nom générique des alliages de cuivre et d’étain) et du fer influença durablement et en profondeur certaines sociétés. Durant l'âge de fer on voit aussi apparaître des verres colorés, composés de SiO2 (principal composé du sable) et d'oxydes métalliques[9].

A : le borane B2H6 est déficient en électrons ;
B : le chlorure de césium a une structure cristalline archétypique ;
C : les ferrocènes sont des complexes organométalliques ;
D : le silicone a de nombreuses utilisations dont les implants mammaires ;
E : le catalyseur de Grubbs a valu le prix Nobel de chimie à son découvreur ;
F : les zéolithes ont des structures poreuses et servent de tamis moléculaires ;
G : l'acétate de cuivre (II) a surpris les théoriciens avec son diamagnétisme.
Les expériences de transmutation permettent aux alchimistes de développer des nouvelles techniques de purification de composés chimiques, comme la distillation, la sublimation ou la cristallisation. De nombreux composés inorganiques sont ainsi isolés, comme le vitriol (acide sulfurique), l'eau-forte (acide nitrique), l'esprit de sel (acide chlorhydrique), le vitriol de lune (sulfate d'argent), les cristaux de Vénus (nitrate de cuivre) ou l'eau régale (mélange d'acide nitrique et d'acide chlorhydrique capable de dissoudre l'or).
En 1675, dans son Cours de chimie, Nicolas Lémery introduisit la distinction entre la « chimie minérale », qui ne faisait intervenir à l'époque que des composés inertes, et la chimie organique, dont les substances sont issues des animaux et des végétaux[10]. Cette distinction, soutenue par la théorie de la force vitale, persista jusqu'au milieu du XIXe siècle. En 1828, l'expérience réalisée par le chimiste Allemand Friedrich Wöhler mit fin à cette démarcation en transformant du cyanate d'ammonium, supposé minéral, en une substance organique, l'urée[11].
Principes
[modifier | modifier le code]Structure atomique
[modifier | modifier le code]Symétrie moléculaire
[modifier | modifier le code]Liaisons moléculaires
[modifier | modifier le code]Composés ioniques
[modifier | modifier le code]Liaison covalente
[modifier | modifier le code]Acides et bases
[modifier | modifier le code]Oxydation et réduction
[modifier | modifier le code]Caractérisation des composés inorganiques
[modifier | modifier le code]Les techniques de caractérisation physico-chimiques des composés inorganiques permettent de déterminer la composition, la structure et les propriétés d'une molécule ou d'un matériau. Plusieurs de ces techniques utilisent l'interaction entre les radiations électromagnétiques et la matière, et ceci sur pratiquement toute la gamme spectrale[12].
- Diffraction : les techniques de diffraction sont particulièrement utilisées pour déterminer les structures des composés chimiques et ont l'avantage d'être des méthodes non destructives. La cristallographie aux rayons X permet de déterminer la position des atomes et des ions qui composent un composé solide. La diffraction de neutrons permet d'obtenir des informations structurales supplémentaires, et en particulier la position des atomes légers.
- Spectroscopies d'absorption : l'absorption (et parfois la ré-émission) des radiations électromagnétiques permet d'obtenir des informations sur les niveaux d'énergie des composés inorganiques et est aussi souvent utilisée pour des analyses quantitatives. Les techniques concernées sont la spectroscopie ultraviolet-visible, la spectroscopie infrarouge et la spectroscopie Raman. Ces techniques sont généralement non destructives.
- Résonance : les techniques de résonance se basent sur l'application d'un champ magnétique pour faire entrer en résonance la séparation entre les niveaux énergétiques d'un composé. La résonance magnétique nucléaire et la résonance paramagnétique électronique impliquent la résonance magnétique, avec les niveaux énergétiques du noyau atomique pour la RMN et des électrons non appariés pour la RPE. La spectroscopie Mössbauer quant à elle est basée sur l'absorption résonante de radiations par le noyau atomique et exploite le fait que les niveaux d'énergie du noyau sont sensibles à l'environnement électronique et nucléaire.
- Ionisation : les techniques d'ionisation permettent de bombarder un échantillon avec des radiations ou des particules de hautes énergies et de mesurer les énergies d'ionisation. Les techniques concernées sont la spectroscopie de photoélectrons, la spectroscopie d'absorption des rayons X et la spectrométrie de masse.
- Analyses chimiques : ces méthodes permettent de déterminer la composition élémentaire des composés et sont généralement destructives. On peut lister dans ces techniques la spectrométrie d'absorption atomique, l'analyse élémentaire, la spectrométrie de fluorescence des rayons X et les méthodes d'analyses thermiques.
- Électrochimie : les techniques électrochimique, et notamment la voltammétrie cyclique, permettent de sonder les réactions d'oxydoréduction d'espèces en solution.
Chimie descriptive des éléments
[modifier | modifier le code]
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | ||||||||||||||||
| 1 | H | He | |||||||||||||||||||||||||||||||
| 2 | Li | Be | B | C | N | O | F | Ne | |||||||||||||||||||||||||
| 3 | Na | Mg | Al | Si | P | S | Cl | Ar | |||||||||||||||||||||||||
| 4 | K | Ca | Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn | Ga | Ge | As | Se | Br | Kr | |||||||||||||||
| 5 | Rb | Sr | Y | Zr | Nb | Mo | Tc | Ru | Rh | Pd | Ag | Cd | In | Sn | Sb | Te | I | Xe | |||||||||||||||
| 6 | Cs | Ba | La | Ce | Pr | Nd | Pm | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | Lu | Hf | Ta | W | Re | Os | Ir | Pt | Au | Hg | Tl | Pb | Bi | Po | At | Rn | |
| 7 | Fr | Ra | Ac | Th | Pa | U | Np | Pu | Am | Cm | Bk | Cf | Es | Fm | Md | No | Lr | Rf | Db | Sg | Bh | Hs | Mt | Ds | Rg | Cn | Nh | Fl | Mc | Lv | Ts | Og | |
| 8 | 119 | 120 | * | ||||||||||||||||||||||||||||||
| * | 121 | 122 | 123 | 124 | 125 | 126 | 127 | 128 | 129 | 130 | 131 | 132 | 133 | 134 | 135 | 136 | 137 | 138 | 139 | 140 | 141 | 142 | |||||||||||
Éléments du groupe principal
[modifier | modifier le code]Hydrogène
[modifier | modifier le code]L'hydrogène est l'élément le plus abondant dans l'univers et le troisième dans la croûte terrestre[13]. La forme stable de l'hydrogène est le dihydrogène, H2, qui n'est présent que sous forme de traces dans la troposphère (0,5 ppm)[14]. Le dihydrogène peut être produit naturellement comme produit de fermentation ou comme sous-produit de la biosynthèse de l'ammoniac[14]. La configuration électronique de l'hydrogène, 1s1, est similaire aux configurations électroniques de valence des métaux alcalins (ns1)[15]. L'élément existe principalement sous trois formes isotopiques : l'hydrogène léger ou protium (1H), le deutérium (2H ou D) et le tritium (3H ou T)[16].
Les propriétés de l'hydrogène sont très diverses, et il peut être une base forte de Lewis, avec l'ion hydrure H−, ou un acide fort de Lewis, avec le proton H+. Ses propriétés chimiques sont riches et variées, et il peut former des composés chimiques avec pratiquement tous les autres éléments. On peut regrouper les composés de l'hydrogène en trois classes principales[17] : les hydrures covalents (formés avec des éléments du bloc p, comme CH4, NH3 ou H2O) ; les hydrures ioniques (formés avec les éléments les plus électropositifs, comme LiH ou CaH2) ; les hydrures interstitiels des métaux de transition (formés avec les de nombreux éléments du bloc d et f). On peut aussi ajouter une dernière classe pour les hydrures dans les complexes métalliques et les complexes du dihydrogène, qui jouent des rôles importants dans la catalyse et l'activation du dihydrogène[18].
Métaux alcalins
[modifier | modifier le code]Les métaux alcalins sont les éléments chimiques de la première colonne du tableau périodique des éléments[19], sauf un. Le lithium (Li), le sodium (Na), le potassium (K), le rubidium (Rb), le césium (Cs) et le francium (Fr) font ainsi partie de cette famille, mais pas l'hydrogène[20]. Aucun des éléments du groupe n'existe à l'état d'élément pur dans la nature, on ne les retrouve que sous la forme de composés[21].
De tous les groupes du tableau périodique, les alcalins montrent de façon claire l'effet de l'augmentation de la taille et de la masse sur les propriétés chimiques et physiques[19]. Ainsi, la réactivité des métaux alcalins augmente avec le numéro atomique (Z). Ceci est principalement relié au fait qu'ils peuvent facilement perdre leur unique électron situé sur la couche de valence et former ainsi des cations chargés +1, la configuration électronique du cation rejoignant celle des gaz rares[22]. L'énergie d'ionisation diminue lorsque l'on descend dans le groupe, et il est donc de plus en plus facile d'éliminer l'électron de valence quand Z augmente, cet électron pouvant participer à des réactions chimiques.
Les métaux alcalins ont tendance à former presque exclusivement des liaisons ioniques avec des éléments non métalliques et halogènes. On les retrouve donc principalement sous forme de sels ou d'oxydes[23].
Les métaux
[modifier | modifier le code]La réduction chimique des cations alcalins pour obtenir la forme métallique est difficile, et la méthode de préparation la plus communément employée pour leur préparation est l'électrolyse des sels fondus du composé chloré[24],[25]. Les métaux alcalins sont très réducteurs, et la réactivité augmente lorsque le numéro atomique augmente[26]; le dioxygène, le dichlore et le dihydrogène peuvent ainsi être réduits facilement. La forme métallique des alcalins doit être conservée dans l'huile afin d'éviter toute réaction avec le dioxygène. Ils réagissent aussi violemment avec l'eau pour former du dihydrogène et des hydroxydes alcalins[24]. Le lithium est le moins réactif en présence d'eau, alors que le sodium réagit violemment, le potassium s'enflamme et le rubidium et le césium explosent[26].
Les métaux alcalins sont solubles dans l'ammoniac liquide et donnent une intense coloration bleue à la solution[27]. Ces solutions sont conductrices, et la couleur et ces propriétés conductrices sont expliquées par la solvatation de l'électron de valence du métal. Dans de l'ammoniac pur, le temps de vie de l'électron solvaté peut être assez long (de l'ordre de 1 % de décomposition par jour)[27]. Ces solutions sont communément utilisées pour préparer divers composés organiques et inorganiques[28].
Les halogènes
[modifier | modifier le code]Les métaux alcalins font partie des éléments les plus électropositifs du tableau périodique, et tendent à former des liaisons ioniques avec les éléments les plus électronégatifs, les halogènes, pour former des sels. Lorsque l'on descend dans le groupe, l'enthalpie de formation devient moins négative pour les fluorures, mais plus négative pour les chlorures, bromures et iodures[29].
Les halogénures de métaux alcalins sont des solides incolores à haut point de fusion qui peuvent être facilement préparés à partir de l'hydroxyde (MOH) ou du carbonate (M2CO3) correspondant, en présence d'un acide halogénohydrique (HX), et suivi d'une recristallisation[30]. Hormis pour le LiF, tous les halogénures de métaux alcalins sont solubles dans l'eau[31]. De très grandes quantités de NaCl et de KCl sont disponibles naturellement sur Terre, et peuvent être purifiées par cristallisation.
Les oxydes et hydroxydes
[modifier | modifier le code]Les métaux alcalins réagissent violemment avec le dioxygène pour former selon les conditions expérimentales des oxydes (M2O), des peroxydes (M2O2) ou des superoxydes (MO2)[32],[33]. Ces différentes variétés d'oxydes réagissent avec l'eau pour donner le cation M+ et l'ion hydroxyde par une réaction acido-basique de Lewis. Les dérivés ozonures (MO3) peuvent être préparés à partir de l'hydroxyde MOH et de l'ozone O3 à basse température, et les dérivés sesquioxydes (M2O3) par décomposition thermique du superoxyde MO2 correspondant[34]. Le peroxyde de sodium est communément employé dans l'industrie comme agent de blanchiment et comme oxydant puissant[33].
Les hydroxydes alcalins sont incolores et transparents. Ils réagissent avec l'eau pour former l'anion hydroxyde et avec le dioxyde de carbone CO2 pour former l'anion carbonate CO2−
3. Le procédé chlore-alcali permet de produire industriellement l'hydroxyde de sodium NaOH utilisé comme réactif par l'industrie chimique et dans la préparation d'autre composés inorganiques[35].
Les complexes de coordination
[modifier | modifier le code]La stabilité des complexes de coordination diminue selon la séquence Li > Na > K > Rb > Cs[36]. Le lithium et le sodium sont des acides de Lewis durs et forment des interactions coulombiennes avec des bases de Lewis dures, comme des ligands oxygénés ou azotés[37]. Les métaux alcalins forment principalement des complexes stables avec des ligands polydentes macrocycliques, comme des éthers couronnes ou des cryptands[38].
Les composés organométalliques
[modifier | modifier le code]Les composés organométalliques de métaux alcalins réagissent rapidement avec l'eau et sont pyrophoriques[39]. Le groupe des organolithiens est le plus important de tous les composés organométalliques du groupe 1, et les organolithiens sont utilisés en synthèse organique comme nucléophiles[40]. Les alkyllithiums sont aussi utilisés dans l'industrie pour la polymérisation des alcènes pour fabriquer le caoutchouc synthétique[39]. Un grand nombre de composés organoalcalins peuvent être préparés en combinant des métaux alcalins avec divers non-métaux[41]. Ces composés sont des bases fortes, et ont d'importantes applications en chimie de synthèse, notamment pour la préparation de composés organométalliques[41],[42].
Métaux alcalino-terreux
[modifier | modifier le code]Les métaux alcalino-terreux sont les éléments chimiques de la deuxième colonne du tableau périodique des éléments[43]. Le béryllium (Be), le magnésium (Mg), le calcium (Ca), le strontium (Sr), le baryum (Ba) et le radium (Ra) font donc partie de cette famille. Tous ces éléments sont de couleur argentée sous leur forme métallique. Le modèle ionique s'applique généralement pour décrire les liaisons dans les composés chimiques des alcalino-terreux[44]. Par certains aspects, le béryllium s'apparente à un métalloïde avec un certain degré de covalence dans ses liaisons avec d'autres éléments[43]. Du fait de la facilité avec laquelle les atomes de ces éléments perdent leurs deux électrons de valence, le degré d'oxydation des alcalino-terreux dans les composés chimiques est +2[45]. Ce sont des réducteurs puissants, mais néanmoins plus faibles que les alcalins.
Groupe 13
[modifier | modifier le code]Les éléments du groupe 13 sont les éléments chimiques de la treizième colonne du tableau périodique des éléments[46]. Le bore (B), l'aluminium (Al), le gallium (Ga), l'indium (In) et le thallium (Tl) font donc partie de cette famille et ces éléments ont diverses propriétés chimiques et physiques.
On peut considérer que le bore est à part dans cette famille[47] car il peut former des composés de type cluster ou des composés polymériques impliquant des atomes d'hydrogène, des métaux et du carbone[48]. Le caractère métallique des éléments augmente lorsque l'on descend dans la colonne du fait de l'augmentation du rayon ionique et de la diminution de l'énergie d'ionisation[46]. Il en résulte que le caractère covalent des liaisons des composés du groupe 13 diminue du bore au thallium, et que donc le caractère ionique des liaisons augmente. Tous ces éléments peuvent former des hydrures, des oxydes et des halogénures à l'état d'oxydation +3[49]. L'état +1 devient stable en descendant dans la colonne, plus particulièrement avec le thallium.
Groupe 14
[modifier | modifier le code]
Le carbone (C), le silicium (Si), le germanium (Ge), l'étain (Sn) et le plomb (Pb) sont des éléments de la 14e colonne du tableau périodique[50]. Les propriétés physico-chimiques de ces éléments varient fortement, le carbone étant considéré comme un non-métal, de même que le silicium, le germanium étant un métalloïde et l'étain et le plomb se rangeant dans la famille des métaux[51]. Cette discontinuité dans la colonne peut être comprise en regardant l'augmentation du rayon atomique et la diminution de l'énergie d'ionisation lorsque l'on passe du carbone au plomb.
La chimie du carbone est principalement développée dans la chimie organique. Tous les éléments du groupe 14 peuvent former des composés binaires simples avec l'hydrogène, l'oxygène, les halogènes et l'azote. Le carbone et le silicium peuvent aussi former des composés de type carbure et siliciure avec des métaux[52]. Concernant l'étain et le plomb, on peut noter que les composés organostanniques à base d'étain et les composés organoplombs sont employés comme fongicides et pesticides[53].
Groupe 15
[modifier | modifier le code]Les éléments du groupe 15, ou pnictogènes, sont les éléments chimiques de la quinzième colonne du tableau périodique des éléments[54]. L'azote (N), le phosphore (P), l'arsenic (As), l'antimoine (Sb) et le bismuth (Bi) font donc partie de cette famille. Les propriétés chimiques des éléments du groupe 15 varient fortement, et notamment entre l'azote et ses congénères car celui-ci possède généralement une coordinence plus faible et est le seul à exister sous forme de molécule diatomique gazeuse dans les conditions normales de température et de pression[54]. Les propriétés de ces éléments sont donc plus difficiles à rationaliser en termes de rayons atomiques et de configurations électroniques. Bien que trouvé communément avec le phosphore, l'arsenic et l'antimoine, l'état d'oxydation +5 ne peut être atteint pour l'azote qu'avec l'oxygène et le fluor. Pour le bismuth c'est l'état d'oxydation +3 qui est le plus stable.
Les éléments du groupe 15 forment des composés binaires avec de nombreux éléments. Les composés azotés sont abondants, et on peut retrouver dans cette famille les nitrures N3−, les hydrures d'azote (comme le NH3, le N2H4 ou le NH2OH), les oxydes d'azote (comme le N2O ou le NO) ou les oxoacides (comme le ONOOH ou le HNO2)[55]. Pratiquement tous les composés azotés peuvent aussi servir de ligands[56].
Chalcogènes
[modifier | modifier le code]Halogènes
[modifier | modifier le code]Gaz nobles
[modifier | modifier le code]
Les gaz nobles sont les éléments chimiques de la dix-huitième colonne du tableau périodique des éléments[57]. L'hélium (He), le néon (Ne), l'argon (Ar), le krypton (Kr), le xénon (Xe) et le radon (Rn) font donc partie de cette famille, et existent à l'état naturel sous forme de gaz monoatomiques. Ils forment une famille d'éléments a priori très peu réactifs car, ayant une couche de valence complète, ils n’ont aucun électron de valence pour former une liaison chimique. Il en découle que ces éléments ont une énergie d'ionisation élevée et une affinité électronique pratiquement nulle[58].
Du fait de cette faible réactivité, peu de composés inorganiques ont pu être caractérisés jusqu'à présent. Depuis les années 1960, il a été démontré que le xénon forme des complexes de coordination stables avec le fluor et l'oxygène[59], le premier complexe isolé étant l'hexafluoroplatinate de xénon Xe+(PtF6)− en 1962[60]. La grande majorité des composés de gaz nobles synthétisés l'ont été à partir du xénon.
Métaux de transition
[modifier | modifier le code]Première série
[modifier | modifier le code]Deuxième et troisième séries
[modifier | modifier le code]Lanthanides
[modifier | modifier le code]Actinides
[modifier | modifier le code]La famille des actinides est composée de 15 éléments chimiques, comprenant l'actinium (Ac), le thorium (Th), le protactinium (Pa), l'uranium (U), le neptunium (Np), le plutonium (Pu), l'américium (Am), le curium (Cm), le berkélium (Bk), le californium (Cf), l'einsteinium (Es), le fermium (Fm), le mendélévium (Md), le nobélium (No) et le lawrencium[61]. La famille tire son nom de l'actinium, le premier élément de la famille. La radioactivité associée avec les actinides n'a pas pu permettre des études très poussées sur cette famille, et les éléments de la fin de la famille ne sont disponibles qu'en très faibles quantités[62]. Cependant, les éléments du début de la famille, et plus particulièrement l'uranium et le plutonium, sont d'une grande importance du fait de leur utilisation dans les centrales nucléaires.
Les propriétés chimiques des actinides ne montrent pas la même uniformité que l'on peut trouver chez les lanthanides tout au long de la famille. De même que les lanthanides, les actinides possèdent de grands rayons atomiques et ioniques, ce qui permet à ces éléments d'offrir une coordinence élevée[63]. Les actinides sont très réactifs vis-à-vis des halogènes et des chalcogènes. Les nucléides communs du thorium et de l'uranium ne présentent que de faibles niveaux de radioactivité, ainsi leurs propriétés chimiques ont pu être bien étudiées[64]. Le cation uranyle forme notamment des complexes avec de nombreux ligands donneurs.
Catalyse
[modifier | modifier le code]Un catalyseur est une substance qui augmente la vitesse d'une réaction sans être lui-même consommé[65]. Les catalyseur sont largement utilisés dans la nature, dans l'industrie et dans les laboratoires. La production industrielle d'acide sulfurique ou d'ammoniac passent toutes deux par exemple par des processus catalytiques. Les catalyseurs jouent aussi un rôle dans la dépollution, comme dans le cas des pots catalytiques des véhicules, ou dans l'amélioration des processus industriels. On peut noter que dans l'industrie, les catalyseurs utilisés sont pratiquement tous de nature inorganique.
Deux types de catalyse peuvent être envisagés : homogène ou hétérogène. Dans le cas de la catalyse homogène, le catalyseur et les réactifs ne forment qu'une seule phase, alors que dans le cas de la catalyse hétérogène, le catalyseur se trouve dans une autre phase que les réactifs et les produits de la réaction catalysée. Les deux formes ont leurs avantages et leurs inconvénients : pour la catalyse hétérogène le catalyseur peut être facilement séparé des réactifs et des produits, mais les réactions ont tendance à nécessiter des hautes températures et pressions et ont une faible sélectivité; inversement, pour la catalyse homogène les réactions peuvent être conduites à plus basses températures et pressions et sont plus sélectives, mais la séparation du catalyseur et des réactifs et des produits en fin de réaction est un inconvénient majeur[66]. Plusieurs réactions peuvent être catalysées en phase homogène, comme la métathèse des alcènes, l'hydrogénation des alcynes, l'hydroformylation ou la carbonylation du méthanol. Les réactions pouvant être catalysées en phase hétérogène sont par exemple l'hydrogénation, la synthèse de l'ammoniac ou la conversion du monoxyde de carbone et de l'hydrogène en hydrocarbures.
Chimie du solide et des matériaux
[modifier | modifier le code]Nanomatériaux, nanoscience et nanotechnologie
[modifier | modifier le code]Un nanomatériau est un matériau dont la dimension est de l'ordre de 1 à 100 nm, ou de façon plus exclusive un matériau de cette dimension qui montre des propriétés absentes à la fois à l'échelle moléculaire et à l'état massif[67]. Les nanomatériaux sont utilisés depuis plusieurs siècles, comme la fabrication d'or colloïdal pour produire un colorant rouge utilisé notamment pour teinter le verre[68]. L'étude des nanomatériaux a particulièrement bénéficié du développement de la microscopie à sonde locale dans les années 1980 qui permet d'observer la matière à l'échelle de l'atome. La chimie inorganique est omniprésente dans les nanotechnologies, que ce soit par exemple dans les nanoparticules métalliques ou les quantum dots. Différents précédés de synthèse peuvent être employés, comme les approches top-down et bottom-up, ou les dépôts chimique ou physique en phase vapeur.
Chimie inorganique industrielle
[modifier | modifier le code]La chimie inorganique industrielle est une branche importante de l'industrie chimique et regroupe de nombreuses applications diverses et variées, comme les engrais minéraux, les matériaux de construction ou les verres et l'émail[69]. De plus, de nombreux produits de base de l'industrie chimique organique, comme les acides minéraux, les alcalins, les agents d'oxydation et les composés halogénés sont préparés dans par cette branche industrielle. Les développements récents des circuits intégrés ou des fibres optiques sont de même dus à l'industrie chimique inorganique.
Chimie inorganique des systèmes biologiques
[modifier | modifier le code]La chimie bioinorganique s'intéresse à l'étude des espèces métalliques dans les systèmes biologiques. Les organismes vivants exploitent les propriétés chimiques et biologiques liées aux interactions entre les ions métalliques et les ligands biologiques de façons très variées, notamment concernant la catalyse enzymatique, la signalisation cellulaire ou la régulation de l'expression des gènes.
Transport
[modifier | modifier le code]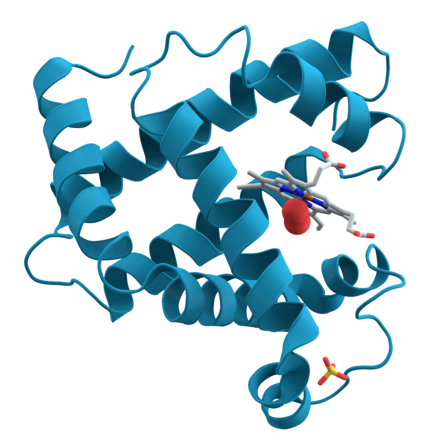
La fixation, le transport et l'utilisation de la molécule de dioxygène O2 chez les organismes vivants sont assurés par des métalloprotéines possédant des cofacteurs inorganiques. Chez la plupart des animaux et plantes l'hémoglobine capte le dioxygène dans les poumons (ou dans les branchies) et le transporte vers les tissus où son stockage est assuré par la myoglobine[71],[72]. Ces deux protéines sont appelées hémoprotéines car elles contiennent un cofacteur de type hème, qui permet la fixation du dioxygène. Les mollusques et les arthropodes utilisent quant à eux une protéine à cuivre, l'hémocyanine, incolore en l'absence de O2 mais de couleur bleue intense en présence de O2[73]. Plus rare, l'hémérythrine est un cofacteur dinucléaire de fer non hémique que l'on retrouve chez certains vers marins[73].
Le flux d'ions K+ et Na+ à travers les membranes est assuré par des canaux ioniques[74],[75]. Ces canaux sont des protéines membranaires et sont responsables de la conduction électrique dans le système nerveux et fonctionnent de manière passive. La pompe sodium-potassium est une protéine transmembranaire qui utilise la dégradation de l'adénosine triphosphate (ATP) en adénosine diphosphate (ADP) pour contrôler la concentration en ion K+ et Na+ à l'intérieur et à l'extérieur de la cellule. Cette protéine joue rôle essentiel dans le maintien du potentiel électrochimique de membrane.
L'assimilation du fer par les organismes est difficile du fait de l'insolubilité du Fe(III), qui est l'état d'oxydation stable trouvé dans la plupart des minéraux[76]. L'évolution a développé des systèmes chimiques complexes pour la capture et la régulation du fer dans l'organisme. La capture implique des ligands appelés sidérophores. Le fer est ensuite transporté par les transferrines et stocké dans les ferritines, deux protéines non hémiques.
Transfert électronique
[modifier | modifier le code]Trois types de métalloprotéines peuvent être identifiés pour les transferts électroniques dans les systèmes biologiques[77]. Les cytochromes sont des hémoprotéines impliquées dans le transfert des électrons dans la chaîne respiratoire mitochondriale et sont constitués d'une porphyrine complexée avec des cations métalliques de fer ou de cuivre[78]. Les protéines fer-soufre possèdent des clusters fer-soufre dans leur structure et participent aussi à des réactions d'oxydo-réduction de la chaîne respiratoire. Le troisième type de protéine est la famille des protéines bleues à cuivre, dont fait partie la plastocyanine qui intervient lors de la photosynthèse. Il faut aussi noter que les transferts d'électrons dans les protéines sont couplés à des processus chimiques comme des transferts d'ions, et notamment des transferts de protons.
Processus catalytiques
[modifier | modifier le code]Les enzymes assurent la catalyse dans les systèmes biologiques en contrôlant non seulement la vitesse de la réaction, mais aussi en favorisant certaines géométries au niveau de leur état de transition grâce à la structure tertiaire de la chaîne polypeptidique[79]. La catalyse enzymatique est indispensable aux organismes vivants pour l'accélération spécifique des réactions nécessaires à leur métabolisme et à la biosynthèse des biomolécules qui les composent. La protéine compose la structure de base des enzymes (apoenzyme), à laquelle il faut ajouter un groupement prosthétique qui peut être un ion métallique complexé ou non.

La catalyse acido-basique par les enzymes permet de produire localement des ions hydroxydes HO− ou hydronium H3O+ dans des conditions de pH qui ne sont que rarement atteintes par les systèmes biologiques[80]. Les organismes vivants utilisent de façon majoritaire le zinc pour ce type de catalyse, car c'est un métal abondant, difficilement oxydable ou réductible, qui forme des liaisons fortes avec les acides aminés, et qui peut lier des ligands exogènes comme H2O. Des exemples de ce type d'enzyme sont l'anhydrase carbonique, la carboxypeptidase ou la phosphatase alcaline[81]. Le manganèse (dans la Rubisco) ou le fer (dans la phosphatase acide ou l'aconitase) sont aussi employés dans la catalyse acido-basique[82].
Un certain nombre d'enzymes permettent aussi d'activer des petites molécules à base d'oxygène comme H2O, H2O2 ou O2[83]. Les peroxydases décomposent les peroxydes, et par exemple la peroxydase de raifort ou la cytochrome c peroxydase possèdent un groupement prosthétique hèminique pour catalyser cette réaction. Les oxydases catalysent la réduction du dioxygène O2 en eau ou en peroxyde d'hydrogène H2O2. La cytochrome c oxydase possède des groupements prosthétiques à base de fer et de cuivre, et cette enzyme est à la base de toutes les formes de vie supérieures. Les oxygénases (comme les cytochromes P450) catalysent l'insertion d'atomes d'oxygène dans des substrats organiques. Enfin la production enzymatique de O2 par photosynthèse implique de nombreuses métalloenzymes, et notamment le photosystème II dans lequel l'eau est oxydée en dioxygène grâce à un site catalytique complexe composé d'atomes de manganèse et d'un atome de cuivre.
Le diazote N2 et le dihydrogène H2 peuvent être réduits par des enzymes à base de sulfure de fer[84]. La nitrogénase transforme le N2 en NH3, et son site actif est composé d'un cluster de sulfure de fer et d'un atome de molybdène (qui peut être remplacé dans certains cas par du vanadium ou du fer). La famille des hydrogénases permet la réduction réversible du H2 en H+, et le site actif est composé de soufre et de fer pour la [FeFe]-hydrogénase, et de soufre, de fer et de nickel pour la [NiFe]-hydrogénase.
Cycles biogéochimiques
[modifier | modifier le code]L'assimilation de certains éléments chimiques par la biosphère fait partie des cycles biogéochimiques. Ces cycles sont des processus de transport et de transformation des éléments chimiques entre la géosphère, l'atmosphère, l'hydrosphère et la biosphère. Au niveau des organismes vivants, ces cycles font intervenir des métalloprotéines capables de fixer, transporter et transformer ces éléments chimiques de façon efficace. Un cas d'école est le cycle de l'azote, qui permet notamment la fixation du diazote N2 atmosphérique par des bactéries présentes dans les sols, et qui implique des enzymes contenant du fer, du cuivre et du molybdène[85].
Biominéralisation
[modifier | modifier le code]Dans la nature, les organismes biologiques produisent des tissues minéralisés, comme les os, les dents ou les coquilles[86]. La biominéralisation est le processus de production de ces minéraux inorganiques par les organismes vivants. Par exemple, le carbonate de calcium CaCO3, sous la forme de calcite ou aragonite, est présent dans les coquilles et les coquilles d'œuf[87], le phosphate de calcium, sous la forme d'hydroxyapatite, est lui le principal composant des os et des dents[88], et les bactéries magnétotactiques synthétisent des cristaux d'oxydes de fer de type magnétite ou de sulfures de fer de type greigite Fe3S4 pour leurs propriétés magnétiques afin de s'orienter et de se déplacer le long de lignes d'un champ magnétique[89].
Médecine
[modifier | modifier le code]Les complexes inorganiques et chélatants jouent en rôle important en pharmacologie. Un des principaux défis dans ce domaine est la détermination du mode d'action du médicament à l'échelle moléculaire, en gardant à l'esprit qu'un médicament administré ne correspond pas à l'espèce réactive au niveau du site actif, et ceci est particulièrement vrai pour les complexes métalliques qui sont souvent plus sensibles à l'hydrolyse que les molécules organiques. Certains métaux non présents dans les organismes vivants comme le platine, l'or, le ruthénium ou le bismuth peuvent aussi avoir des effets pharmacologiques[90]. La chimie inorganique intervient à plusieurs niveaux en pharmacologie, comme pour le traitement d'excès de fer dans l'organisme par des ligands inspirés des sidérophores[91], le traitement de certains cancers par le cisplatine (cis-[PtCl2(NH3)2]) comme inhibiteur de la réplication de l'ADN[92], l'utilisation de complexes d'or contre la polyarthrite rhumatoïde[93], ou l'utilisation de complexes métalliques à base de gadolinium comme agents de contraste en imagerie médicale[94].
Toxicité et pollution
[modifier | modifier le code]Éléments-traces métalliques
[modifier | modifier le code]La notion d’éléments-traces métalliques tend à remplacer celle de métaux lourds qui a été et qui reste un concept mal défini car associant des métaux toxiques réellement lourds à d'autres l'étant moins. Une partie de ces éléments-traces métalliques est toxique, ou toxique au-delà d'un certain seuil, ou radioactive (radionucléides). Les métaux diffèrent des composés toxiques organiques dans le sens où ils ne peuvent être dégradés en forme non toxiques, bien qu'ils peuvent ultimement être transformés en formes insolubles et de fait ne plus être disponible biologiquement[95]. Les éléments-traces métalliques se retrouvent dans l'air, dans l'eau et dans les sols.
On considère principalement cinq éléments qui présentent les dangers les plus importants pour l'environnement du fait de leur toxicité et de leur distribution à grande échelle : le mercure (Hg), le plomb (Pb), le cadmium (Cd), le chrome (Cr) et l'arsenic (As)[96]. La notion de spéciation chimique est importante de ce point de vue, car elle permet de distinguer les différentes formes possibles de chaque élément dans un environnement donné[97]. Ces éléments ne sont pas particulièrement toxiques dans leur forme métallique (M°), à l'exception notable des vapeurs de mercure qui sont hautement toxiques. Les formes cationiques de ces métaux sont elles dangereuses, et notamment lorsqu'elle sont liées à des courtes chaines carbonées[98]. On peut aussi noter la forte affinité de ces cations pour le soufre, qui est présent dans de nombreuses enzymes contrôlant des réactions métaboliques dans le corps humain. La liaison métal-soufre modifie l'activité de ces enzymes, ce qui conduit à des troubles de la santé chez les êtres vivants.
Certaines substances peuvent aussi présenter des phénomènes de biomagnification : les taux des éléments-traces métalliques croissent à chaque stade du réseau trophique (chaîne alimentaire)[99]. Ceci concerne principalement le mercure. De nombreuses espèces aquatiques peuvent bioconcentrer des métaux, comme les huîtres et les moules qui peuvent présenter des niveaux de mercure ou de cadmium 100 000 fois supérieurs à ceux de l'eau dans laquelle elles vivent[98]. Ainsi, la majeure partie des métaux ingérés par l'homme provient de la nourriture plutôt que de l'eau potable.
Pollution de l'air
[modifier | modifier le code]
Les oxydes d'azote et de soufre sont des polluants majeurs de l'atmosphère. Les oxydes d'azote NOx sont principalement produits lors de la combustion de combustibles fossiles à haute température[100] et les chauffages au bois génèrent plus d'émissions par unité d'énergie produite que les installations de combustion fonctionnant aux énergies fossiles[101]. La houille de mauvaise qualité et le pétrole contiennent des composés soufrés et génèrent du dioxyde de soufre SO2 lors de leur combustion, mais le dioxyde de soufre dans l'atmosphère provient principalement des éruptions volcaniques[102]. Les pluies acides sont les résultantes directes ces deux familles de polluants[103].
De nombreux centres urbains subissent des épisodes de pollution atmosphérique du fait de niveaux d'ozone très importants, phénomène connu sous le nom de smog[104]. L'ozone troposphérique O3, c'est-à-dire de l'ozone des basses couches de l'atmosphère, est un polluant secondaire. Il n'est pas émis directement dans l'air mais résulte de réactions photochimique complexes impliquant des précurseurs, essentiellement des oxydes d'azote issus des gaz d'échappement des voitures. Le smog n'est pas le seul problème de pollution atmosphérique créé par les automobiles. D'autres polluants, comme le plomb, proviennent de la combustion de l'essence[105]. Le plomb sous la forme tétraéthylplomb a été utilisé comme agent antidétonant en augmentant l'indice d'octane. Le développement de l'essence sans plomb dans les années 1970 a permis de réduire les nuisances dues aux intoxications au plomb.
Pollution de l'eau et des sols
[modifier | modifier le code]Les polluants inorganiques de l'eau et des sols proviennent principalement des activités humaines, comme les engrais minéraux à base de phosphates (PO3−
4) et de nitrates (NO−
3)[106], ou les éléments-traces métalliques dégagés par des drainages miniers acides [107], les ruissellements urbains et les activités industrielles[108].
Pollution radioactive
[modifier | modifier le code]La pollution radioactive résulte d'un épandage, d'un accident ou d'une explosion nucléaire au cours desquels des radioisotopes sont répandus. Ces radioisotopes peuvent être d'origine naturelle, comme pour l'uranium 238, l'uranium 235 ou le thorium 232, ou dus à l'activité humaine, comme pour les déchets radioactifs provenant de laboratoires de recherche, industriels ou médicaux, les déchets provenant de la production d'énergie nucléaire, ou les bombes nucléaires et les explosions nucléaires expérimentales[109]. La pollution radioactive est nocive pour l'homme car les radioisotopes peuvent se désintégrer en émettant des rayonnements ionisants qui endommagent les constituants cellulaires des organismes vivants.
Littérature en chimie inorganique
[modifier | modifier le code]Livres de référence
[modifier | modifier le code]- (en) P. W. Atkins, T. Overton et al., Inorganic chemistry, New York (États-Unis), W. H. Freeman and Company, , 5e éd., 830 p. (ISBN 978-0-19-923617-6, présentation en ligne).
- (en) C. E. Housecroft et A. G. Sharpe, Inorganic chemistry, Harlow (Royaume-Uni), Prentice Hall, , 2e éd., 992 p. (ISBN 978-0-13-039913-7, présentation en ligne).
- (en) P. A. Cox, Inorganic chemistry, BIOS Scientific Publishers, , 2e éd., 359 p. (ISBN 978-1-85996-289-3, présentation en ligne).
- (en) K. H. Büchel, H.-H. Moretto et al., Industrial inorganic chemistry, Weinheim (Allemagne), Wiley-VCH, , 2e éd., 667 p. (ISBN 978-3-527-29849-5, présentation en ligne).
- (en) F. A. Cotton, G. Wilkinson et al., Advanced inorganic chemistry, John Wiley & Sons, , 6e éd., 1376 p. (ISBN 978-0-471-19957-1, présentation en ligne).
- (en) N. N. Greenwood et A. Earnshaw, Chemistry of the elements, Oxford/Boston, Butterworth-Heinemann, , 2e éd., 1359 p. (ISBN 978-0-7506-3365-9, présentation en ligne).
- J. E. Huheey, E. A. Keiter et al. (trad. André Pousse et Jean Fischer), Chimie inorganique [« Inorganic chemistry: Principles of structure and reactivity »], De Boeck Université, , 964 p. (ISBN 978-2-8041-2112-9, présentation en ligne, lire en ligne).
Publications scientifiques
[modifier | modifier le code]Les sources primaires en chimie inorganique se résument à une quarantaine de revues scientifiques selon le Journal Citation Reports[110], auxquelles il faut ajouter une dizaine de revues généralistes en chimie, comme Angewandte Chemie, Chemical Communications ou Journal of the American Chemical Society. Quelques articles paraissent aussi chaque année dans les revues scientifiques généralistes Nature et Science. Le tableau suivant présente les principales revues scientifiques internationales spécialisées en chimie inorganique.
| Titre | Éditeur | Année de création | Facteur d'impact[110] (2015) |
|---|---|---|---|
| Applied Organometallic Chemistry | John Wiley & Sons | 1987 | 2,452 |
| Coordination Chemistry Reviews | Elsevier Science | 1973 | 12,994 |
| Dalton Transactions | Royal Society of Chemistry | 1966 | 4,177 |
| European Journal of Inorganic Chemistry | John Wiley & Sons | 1998 | 2,686 |
| Inorganic Chemistry | American Chemical Society | 1962 | 4,820 |
| Inorganic Chemistry Communications | Elsevier Science | 1998 | 1,762 |
| Inorganica Chimica Acta | Elsevier Science | 1967 | 1,918 |
| Journal of Biological Inorganic Chemistry | Springer Verlag | 1996 | 2,495 |
| Journal of Inorganic Biochemistry | Elsevier Science | 1971 | 3,205 |
| Journal of Organometallic Chemistry | Elsevier Science | 1963 | 2,336 |
| Journal of Solid State Chemistry | Elsevier Science | 1969 | 2,265 |
| Organometallics | American Chemical Society | 1982 | 4,186 |
| Polyhedron | Elsevier Science | 1955 | 2,108 |
| Zeitschrift für Anorganische und Allgemeine Chemie | John Wiley & Sons | 1892 | 1,261 |
Les sources secondaires sont les publications scientifiques publiant des articles de revue en chimie inorganique, principalement Coordination Chemistry Reviews, Chemical Society Reviews et Chemical Reviews. Certaines des publications primaires présentant des résultats scientifiques proposent aussi dans chaque numéro des articles de revue. La collection Inorganic Syntheses est une série de livres éditée depuis 1939 qui vise à publier les procédures détaillées de synthèses de composés inorganiques[111].
Prix Nobel de chimie liés à la chimie inorganique
[modifier | modifier le code]| Année | Lauréat(s) | Nationalité | Travaux récompensés |
|---|---|---|---|
| 1904 | William Ramsay | En reconnaissance de la découverte dans l'air d'éléments gazeux inertes, et de la détermination de leur position dans le tableau périodique | |
| 1906 | Henri Moissan | En reconnaissance des grands services qu'il a rendus par la découverte du fluor et de ses propriétés, et pour avoir mis à la disposition de la science le four électrique qui porte son nom | |
| 1911 | Marie Curie | Pour les services rendus à l'avancement de la chimie par sa découverte des éléments radium et polonium, pour avoir isolé le radium et étudié la nature et les composés de cet élément remarquable | |
| 1913 | Alfred Werner | Pour ses travaux sur les liaisons des atomes dans les molécules, grâce auxquels il a porté un éclairage nouveau sur des études antérieures et ouvert de nouveaux domaines de recherche tout particulièrement en chimie minérale | |
| 1918 | Fritz Haber | Pour la synthèse de l'ammoniac à partir de ses éléments NB : prix décerné en 1919 et remis en 1920 | |
| 1951 | Edwin McMillan et Glenn Theodore Seaborg |
Pour leurs découvertes dans la chimie des éléments transuraniens | |
| 1954 | Linus Carl Pauling | Pour ses recherches sur la nature de la liaison chimique et leurs applications à la détermination de la structure de substances complexes | |
| 1963 | Karl Ziegler et Giulio Natta |
Pour leurs découvertes dans le domaine de la chimie et de la technologie des hauts polymères | |
| 1964 | Dorothy Crowfoot Hodgkin | Pour la détermination par les techniques des rayons X de la structure d'importantes substances biologiques | |
| 1966 | Robert Sanderson Mulliken | Pour son travail fondamental concernant les liaisons chimiques et la structure électronique des molécules par la méthode des orbitales moléculaires | |
| 1973 | Ernst Otto Fischer et Geoffrey Wilkinson |
Pour leurs travaux de pionniers, réalisés indépendamment, sur les composés organométalliques appelés composés sandwich | |
| 1976 | William Lipscomb | Pour ses travaux sur la structure des boranes, qui ont apporté un nouvel éclairage sur la liaison chimique | |
| 1983 | Henry Taube | Pour ses travaux sur les mécanismes des réactions par transfert d'électrons, en particulier dans les complexes métalliques | |
| 1985 | Herbert Aaron Hauptman et Jerome Karle |
Pour leurs réalisations remarquables dans la mise au point de méthodes directes de détermination des structures cristallines | |
| 1996 | Robert Curl, Richard Smalley et Harold Kroto |
Pour leur découverte des fullerènes | |
| 1998 | Walter Kohn¹ et John A. Pople² |
¹ Pour ses développements de la théorie de la fonctionnelle de la densité ² Pour avoir développé des méthodes de calculs informatiques en chimie quantique | |
| 2001 | William S. Knowles¹, K. Barry Sharpless² et Ryoji Noyori¹ |
¹ Pour leurs travaux sur les réactions d'hydrogénation avec catalyse chirale ² Pour ses travaux sur les réactions d'oxydation en catalyse chirale | |
| 2005 | Yves Chauvin, Robert Grubbs et Richard R. Schrock |
Pour leurs travaux sur le développement de la méthode de la métathèse en synthèse organique | |
| 2010 | Richard Heck, Ei-ichi Negishi et Akira Suzuki |
Pour les réactions de couplage catalysées par le palladium en synthèse organique | |
| 2016 | Jean-Pierre Sauvage James Fraser Stoddart Bernard L. Feringa |
Pour la conception et la synthèse de machines moléculaires |
Notes et références
[modifier | modifier le code]- M. Bernard, Cours de chimie minérale, Éditions Dunod, , 2e éd., 405 p. (ISBN 978-2-10-002067-6, présentation en ligne).
- R. Quelet, Précis de chimie : Chimie minérale, Presses universitaires de France, .
- Monoxyde de carbone (CO), dioxyde de carbone (CO2). D'autres oxydes de carbone, inorganiques et organiques, sont présentés dans un tableau situé dans le bas de page de l’article « oxyde de carbone ».
- Sur les composés carbonés « inorganiques », voir l'article « composé organique ».
- Artificiel ou naturel : Substances naturelles ou synthétiques, sur le projet Wikiversité, consulté le 4 octobre 2014.
- Huheey et Keiter 1996, chap. 15 (« La chimie organométallique »), p. 623.
- Huheey et Keiter 1996, chap. 15 (« Chaînes, cycles cages et clusters inorganiques »), p. 742-750.
- (en) « Careers in Chemistry: Inorganic Chemistry », American Chemical Society
- Miessler et Fischer 2014, chap. 1 (« Introduction to Inorganic Chemistry »), p. 5.
- Nicolas Lémery, Cours de chimie, contenant la manière de faire les opérations qui sont en usage dans la médecine, par une méthode facile, avec des raisonnements sur chaque opération, pour l’instruction de ceux qui veulent s’appliquer à cette science, Paris, L.-C. d'Houry fils, (1re éd. 1675), 782 p. (BNF 37264707, lire en ligne), p. 2.
- (de) Friedrich Wöhler, « Ueber künstliche Bildung des Harnstoffs », Ann. Phys. (Berlin), vol. 88, no 2, , p. 253–256 (ISSN 0003-3804, DOI 10.1002/andp.18280880206, lire en ligne).
- Atkins et Overton 2010, chap. 8 (« Physical techniques in inorganic chemistry »), p. 223.
- Miessler et Fischer 2014, chap. 8 (« Chemistry of the Main Group Elements »), p. 258.
- Atkins et Overton 2010, chap. 10 (« Hydrogen »), p. 274.
- Miessler et Fischer 2014, chap. 8 (« Chemistry of the Main Group Elements »), p. 257.
- Atkins et Overton 2010, chap. 10 (« Hydrogen »), p. 275.
- Atkins et Overton 2010, chap. 10 (« Hydrogen »), p. 276-277.
- Greenwood et Earnshaw 1997, chap. 3 (« Hydrogen »), p. 64-67.
- Cotton et Wilkinson 1999, chap. 3 (« The group 1 elements: Li, Na, K, Rb, Cs, Fr »), p. 92.
- (en) N. G. Connelly, R. M. Hartshorn et al., Nomenclature of inorganic chemistry : IUPAC recommendations, Cambridge (Royaume-Uni), International Union of Pure and Applied Chemistry, , 377 p. (ISBN 0-85404-438-8, présentation en ligne, lire en ligne), chap. 3 (« Elements »), p. 51.
- (en) M. Halka et B. Nordstrom, Alkali and alkaline earth metals, New York (États-Unis), Infobase Publishing, coll. « Periodic Table of the Elements Set », , 192 p. (ISBN 978-0-8160-7369-6, présentation en ligne), xvi.
- Greenwood et Earnshaw 1997, chap. 4 (« Lithium, Sodium, Potassium, Rubidium, Caesium and Francium »), p. 76.
- Halka et Nordstrom 2010, p. xvii.
- (en) « Visual Elements: Group 1 - The Alkali Metals », sur le site de la Royal Society of Chemistry (consulté le ).
- (en) R. B. Heslop et P. L. Robinson, Inorganic chemistry : a guide to advanced study, Amsterdam (Pays-Bas), Elsevier Science, , 2e éd., 591 p., chap. 14 (« The Alkali Metals (Group I A) »), p. 248.
- Cotton et Wilkinson 1999, chap. 3 (« The group 1 elements: Li, Na, K, Rb, Cs, Fr »), p. 95.
- Cotton et Wilkinson 1999, chap. 3 (« The group 1 elements: Li, Na, K, Rb, Cs, Fr »), p. 96.
- Greenwood et Earnshaw 1997, chap. 4 (« Lithium, Sodium, Potassium, Rubidium, Caesium and Francium »), p. 78-79.
- Atkins et Overton 2010, chap. 11 (« The Group 1 elements »), p. 299.
- Greenwood et Earnshaw 1997, chap. 4 (« Lithium, Sodium, Potassium, Rubidium, Caesium and Francium »), p. 82.
- Atkins et Overton 2010, chap. 11 (« The Group 1 elements »), p. 296.
- Atkins et Overton 2010, chap. 11 (« The Group 1 elements »), p. 300.
- Greenwood et Earnshaw 1997, chap. 4 (« Lithium, Sodium, Potassium, Rubidium, Caesium and Francium »), p. 84.
- Greenwood et Earnshaw 1997, chap. 4 (« Lithium, Sodium, Potassium, Rubidium, Caesium and Francium »), p. 85.
- Atkins et Overton 2010, chap. 11 (« The Group 1 elements »), p. 302.
- Greenwood et Earnshaw 1997, chap. 4 (« Lithium, Sodium, Potassium, Rubidium, Caesium and Francium »), p. 90.
- Atkins et Overton 2010, chap. 11 (« The Group 1 elements »), p. 305.
- Huheey et Keiter 1996, chap. 12 (« La chimie de coordination : structures »), p. 525-531.
- Atkins et Overton 2010, chap. 11 (« The Group 1 elements »), p. 307.
- Cotton et Wilkinson 1999, chap. 3 (« The group 1 elements: Li, Na, K, Rb, Cs, Fr »), p. 106.
- (en) K. Ruhlandt-Senge, K. W. Henderson et al., Alkali Metal Organometallics : Structure and Bonding, Compounds of groups 1-2 and 11-12, Elsevier Science, coll. « Comprehensive Organometallic Chemistry » (no 2), , 3e éd., 498 p. (ISBN 978-0-08-044592-2, présentation en ligne), p. 1-65.
- (en) J. D. Dunitz (dir.), P. Hemmerich (dir.) et al., Alkali metal complexes with organic ligands, Springer, coll. « Structure and Bonding » (no 16), , 189 p. (ISBN 978-3-540-06423-7, présentation en ligne).
- Cotton et Wilkinson 1999, chap. 4 (« The group 2 elements: Be, Mg, Ca, Sr, Ba, Ra »), p. 111.
- Atkins et Overton 2010, chap. 12 (« The Group 2 elements »), p. 309.
- Huheey et Keiter 1996, chap. 14 (« Un peu de chimie descriptive des métaux »), p. 594.
- Atkins et Overton 2010, chap. 13 (« The Group 13 elements »), p. 325.
- Cotton et Wilkinson 1999, chap. 5 (« Boron »), p. 131.
- Atkins et Overton 2010, chap. 13 (« The Group 13 elements »), p. 329.
- Atkins et Overton 2010, chap. 13 (« The Group 13 elements »), p. 327.
- Atkins et Overton 2010, chap. 14 (« The Group 14 elements »), p. 350.
- Cotton et Wilkinson 1999, chap. 8 (« The group 14 elements: SI, Ge, Sn, Pb »), p. 258.
- Atkins et Overton 2010, chap. 14 (« The Group 14 elements »), p. 352.
- Atkins et Overton 2010, chap. 14 (« The Group 14 elements »), p. 371.
- Atkins et Overton 2010, chap. 15 (« The Group 15 elements »), p. 375.
- Cotton et Wilkinson 1999, chap. 9 (« Nitrogen »), p. 315-340.
- Cotton et Wilkinson 1999, chap. 9 (« Nitrogen »), p. 340-378.
- Cotton et Wilkinson 1999, chap. 3 (« The group 18 elements: He, Ne, Ar, Kr, Xe, Rn »), p. 586.
- Atkins et Overton 2010, chap. 18 (« The Group 18 elements »), p. 440.
- Greenwood et Earnshaw 1997, chap. 18 (« The Noble Gases: Helium, Neon, Argon, Krypton, Xenon and Radon »), p. 892.
- (en) N. Bartlett, « Xenon hexafluoroplatinate Xe+[PtF6]− », Proc. Chem. Soc., no 6, , p. 218-220 (ISSN 0369-8718, DOI 10.1039/PS9620000197).
- Greenwood et Earnshaw 1997, chap. 31 (« The Actinide and Transactinide Elements (Z = 90 - 112) »), p. 1252.
- Atkins et Overton 2010, chap. 23 (« The f-block elements »), p. 592.
- Atkins et Overton 2010, chap. 23 (« The f-block elements »), p. 593.
- Atkins et Overton 2010, chap. 23 (« The f-block elements »), p. 595.
- Atkins et Overton 2010, chap. 26 (« Catalysis »), p. 690.
- Huheey et Keiter 1996, chap. 15 (« La chimie organométallique »), p. 705-706.
- Atkins et Overton 2010, chap. 25 (« Nanomaterials, nanoscience, and nanotechnology »), p. 653.
- (en) L. B. Hunt, « The true story of purple of Cassius », Gold Bull., vol. 9, no 4, , p. 134-139 (ISSN 0017-1557, DOI 10.1007/BF03215423, lire en ligne).
- (en) K. H. Büchel, H. H. Moretto et al., Industrial inorganic chemistry, Weinheim (Allemagne), Wiley-VCH, , 2e éd., 667 p. (ISBN 978-3-527-29849-5 et 9783527613328, DOI 10.1002/9783527613328, présentation en ligne).
- (en) T. Takano, « Structure of myoglobin refined at 2.0 Å resolution: II. Structure of deoxymyoglobin from sperm whale », J. Mol. Biol., vol. 110, no 3, , p. 569–584 (ISSN 0022-2836, DOI 10.1016/S0022-2836(77)80112-5).
- Crichton et Louro 2013, chap. 1 (« An Overview of the Roles of Metals in Biological Systems »), p. 12-13.
- Huheey et Keiter 1996, chap. 15 (« La chimie inorganique des systèmes biologiques»), p. 891-910.
- Atkins et Overton 2010, chap. 27 (« Biological inorganic chemistry »), p. 740.
- Crichton et Louro 2013, chap. 1 (« An Overview of the Roles of Metals in Biological Systems »), p. 3-5.
- Atkins et Overton 2010, chap. 27 (« Biological inorganic chemistry »), p. 731-732.
- Atkins et Overton 2010, chap. 27 (« Biological inorganic chemistry »), p. 735-738.
- Atkins et Overton 2010, chap. 27 (« Biological inorganic chemistry »), p. 741-745.
- (en) R. Crichton, Biological inorganic chemistry : A new introduction to molecular structure and function, Amsterdam, Elsevier, , 2e éd., 460 p. (ISBN 978-0-444-53782-9, DOI 10.1016/B978-0-444-53782-9.00013-9, lire en ligne), chap. 13 (« Iron: essential for almost all life »), p. 259-262.
- Huheey et Keiter 1996, chap. 15 (« La chimie inorganique des systèmes biologiques»), p. 919.
- Atkins et Overton 2010, chap. 27 (« Biological inorganic chemistry »), p. 746.
- (en) R. Crichton, Biological inorganic chemistry : A new introduction to molecular structure and function, Amsterdam, Elsevier, , 2e éd., 460 p. (ISBN 978-0-444-53782-9, DOI 10.1016/B978-0-444-53782-9.00013-9, lire en ligne), chap. 12 (« Zinc: Lewis acid and gene regulator »), p. 231-232.
- Atkins et Overton 2010, chap. 27 (« Biological inorganic chemistry »), p. 749-751.
- Atkins et Overton 2010, chap. 27 (« Biological inorganic chemistry »), p. 751-759.
- (en) W. Weigand et P. Schollhammer, Bioinspired catalysis : metal-sulfur complexes, Weinheim, Allemagne, Wiley-VCH, , 418 p. (ISBN 978-3-527-33308-0 et 9783527664160, DOI 10.1002/9783527664160, lire en ligne).
- Atkins et Overton 2010, chap. 27 (« Biological inorganic chemistry »), p. 765-767.
- Atkins et Overton 2010, chap. 27 (« Biological inorganic chemistry »), p. 771-772.
- (en) E. Bäuerlein (dir.), J. L. Arias et al., Handbook of biomineralization : biological aspects and structure formation, vol. 1 : Biological aspects and structure formation, Weinheim, Germany, Wiley-VCH, (ISBN 978-3-527-31641-0, DOI 10.1002/9783527619443.ch18, présentation en ligne), chap. 18 (« Eggshell Growth and Matrix Macromolecules »), p. 309-327
- (en) E. Bäuerlein (dir.), K. Kawasaki et al., Handbook of biomineralization : biological aspects and structure formation, vol. 1 : Biological aspects and structure formation, Weinheim, Germany, Wiley-VCH, (ISBN 978-3-527-31641-0, DOI 10.1002/9783527619443.ch19, présentation en ligne), chap. 19 (« Genetic Basis for the Evolution of Vertebrate Mineralized Tissue »), p. 331-347
- (en) E. Bäuerlein (dir.), R. B. Frankel et al., Handbook of biomineralization : biological aspects and structure formation, vol. 1 : Biological aspects and structure formation, Weinheim, Germany, Wiley-VCH, (ISBN 978-3-527-31641-0, DOI 10.1002/9783527619443.ch8, présentation en ligne), chap. 8 (« Magnetic Microstructure of Magnetotactic Bacteria »), p. 127-144
- Atkins et Overton 2010, chap. 27 (« Biological inorganic chemistry »), p. 772-776.
- (en) O. Andersen, « Principles and recent developments in chelation treatment of metal intoxication », Chem. Rev., vol. 99, no 9, , p. 2683–2710 (ISSN 0009-2665, DOI 10.1021/cr980453a).
- (en) E. Wong et C. M. Giandomenico, « Current status of platinum-based antitumor drugs », Chem. Rev., vol. 99, no 9, , p. 2451-2466 (ISSN 0009-2665, DOI 10.1021/cr980420v).
- (en) C. F. Shaw, « Gold-based therapeutic agents », Chem. Rev., vol. 99, no 9, , p. 2589–2600 (ISSN 0009-2665, DOI 10.1021/cr980431o).
- (en) P. Caravan, J. J. Ellison et al., « Gadolinium(III) chelates as MRI contrast agents: structure, dynamics, and applications », Chem. Rev., vol. 99, no 9, , p. 2293–2352 (ISSN 0009-2665, DOI 10.1021/cr980440x).
- Baird et Cann 2008, chap. 15 (« Toxic heavy metals »), p. 664.
- Baird et Cann 2008, chap. 15 (« Toxic heavy metals »), p. 663.
- (en) L. Ebdon, L. Pitts et al., Trace element speciation for environment, Cambridge, Royal Society of Chemistry, , 418 p. (ISBN 978-0-85404-459-7 et 9781847552204, DOI 10.1039/9781847552204, présentation en ligne).
- Baird et Cann 2008, chap. 15 (« Toxic heavy metals »), p. 666.
- (en) R. P. Mason, Trace metals in aquatic systems, Chichester, Wiley-Blackwell, , 440 p. (ISBN 978-1-4051-6048-3 et 9781118274576, présentation en ligne), chap. 8 (« Trace metals and organisms »), p. 406-414.
- Baird et Cann 2008, chap. 3 (« The chemistry of ground-level air »), p. 99.
- Sources de polluants atmosphériques: chauffages au bois, sur le site de l'Office fédéral de l'environnement (Suisse).
- Baird et Cann 2008, chap. 3 (« The chemistry of ground-level air »), p. 95.
- Andrews et Brimblecombe 2004, chap. 3 (« The atmosphere »), p. 55-56.
- Baird et Cann 2008, chap. 3 (« The chemistry of ground-level air »), p. 97.
- Andrews et Brimblecombe 2004, chap. 3 (« The atmosphere »), p. 54.
- Baird et Cann 2008, chap. 14 (« The pollution and purification of water »), p. 620.
- Andrews et Brimblecombe 2004, chap. 5 (« The chemistry of continental waters »), p. 156-159.
- Andrews et Brimblecombe 2004, chap. 5 (« The chemistry of continental waters »), p. 170.
- (en) J. Kónya et N. M. Nagy, Nuclear and radiochemistry, London/Waltham, MA, Elsevier, , 430 p. (ISBN 978-0-12-391430-9, présentation en ligne), chap. 13 (« Environmental radioactivity »), p. 375-378.
- « Journal Citation Reports », sur le site de Thomson Reuters (consulté le ).
- « Inorganic Syntheses », sur le site de John Wiley & Sons (consulté le ).
Voir aussi
[modifier | modifier le code]Bibliographie
[modifier | modifier le code]- Robert Valls, Chimie inorganique. De la classification périodique au cristal, ISTE Editions Limited, , 247 p. (ISBN 9781784053932, lire en ligne)
- (en) G. L. Miessler, P. J. Fischer et al., Inorganic chemistry, Saddle River, NJ (États-Unis), Prentice Hall, , 5e éd., 696 p. (ISBN 978-0-321-81105-9, présentation en ligne).
- (en) R. Crichton et R. Louro, Practical approaches to biological inorganic chemistry, Amsterdam/Waltham, Elsevier, , 317 p. (ISBN 978-0-444-56351-4, présentation en ligne).
- (en) P. W. Atkins, T. Overton et al., Inorganic chemistry, New York (États-Unis), W. H. Freeman and Company, , 5e éd., 830 p. (ISBN 978-0-19-923617-6, présentation en ligne).
- (en) C. Baird et M. C. Cann, Environmental chemistry, New York, W. H. Freeman, , 4e éd., 650 p. (ISBN 978-1-4292-0146-9, présentation en ligne).
- (en) C. E. Housecroft et A. G. Sharpe, Inorganic chemistry, Harlow (Royaume-Uni), Prentice Hall, , 2e éd., 992 p. (ISBN 978-0-13-039913-7, présentation en ligne).
- (en) J. E. Andrews, P. Brimblecombe et al., An introduction to environmental chemistry, Wiley-Blackwell, , 2e éd., 320 p. (ISBN 978-0-632-05905-8, présentation en ligne).
- (en) F. A. Cotton, G. Wilkinson et al., Advanced inorganic chemistry, John Wiley & Sons, , 6e éd., 1376 p. (ISBN 978-0-471-19957-1, présentation en ligne).
- (en) N. N. Greenwood et A. Earnshaw, Chemistry of the elements, Oxford/Boston, Butterworth-Heinemann, , 2e éd., 1359 p. (ISBN 978-0-7506-3365-9, présentation en ligne).
- J. E. Huheey, E. A. Keiter et al. (trad. André Pousse et Jean Fischer), Chimie inorganique [« Inorganic chemistry: Principles of structure and reactivity »], De Boeck Université, , 964 p. (ISBN 978-2-8041-2112-9, présentation en ligne, lire en ligne).
Articles connexes
[modifier | modifier le code]- Nomenclature des composés inorganiques
- Liste de composés inorganiques
- Liste de chimistes inorganiques