Contents
Adult Onset Still’s Disease
Adult Onset Still’s Disease (AOSD) also known as systemic onset juvenile idiopathic arthritis, adult Still’s disease or Wissler-Fanconi syndrome, is a rare polygenetic, multisystem autoimmune (when the body’s immune system attacks its own healthy cells, tissues, and organs) or autoinflammatory disorder (autoinflammatory diseases are a group of disorders characterized by recurrent episodes of inflammation due to abnormal activation of the innate immune system leading recurring episodes of inflammation and fever) characterized by daily high-spiking fever, inflammatory polyarthritis, a transient salmon-pink maculopapular rash, leukocytosis with neutrophilia, elevated ferritin levels 1, 2, 3, 4, 5, 6. Adult onset Still’s disease can affect any joint in your body, but most commonly involved joints are knees, ankle, elbows, shoulders, and fingers 7, 8, 9, 10, 11, 12, 13, 14. Adult-onset Still’s disease (AOSD) begins in adulthood, so it’s compared to rheumatoid arthritis. Inflammation may affect a few joints at first. Over time, more joints may be involved. Some people may have only one bout of the illness followed by lasting remission (no visible symptoms), while others develop chronic arthritis. Adult onset Still’s disease affects men and women equally, usually young adults between the ages of 16 and 35.
The primary clinical features seen in adult-onset Still’s disease are fever, rash, and arthritis or arthralgia. They are seen in about 75 to 95 percent of patients 15. Other common symptoms are muscle pain or discomfort (myalgia), sore throat (pharyngitis), swollen lymph nodes (lymphadenopathy) and enlarged spleen (splenomegaly). Less commonly observed symptoms are enlarged liver (hepatomegaly), pleural lining inflammation (pleuritis), inflammation of the pericardium (the sac-like membrane surrounding the heart), and abdominal pain. Fever is usually quotidian (daily recurring fever with temperature returning to normal between fever spikes) most often occurring late during the day and generally precedes other symptoms. The temperature can change by 4ºC within four hours 16. Approximately 20 percent of cases do not have complete defervescence as fever persists between spikes or an additional spike occur in the morning (double quotidian fever). Adult-onset Still’s disease may also present as fever of unknown origin (FUO), and a temperature of more than 39.5ºC suggests monophasic pattern more strongly 17. A rash is classically evanescent, salmon-colored, macular or maculopapular which is usually not itchy and occurs with the fever. Salmon-colored, macular or maculopapular rash is mainly seen on the trunk and extremities but can also be seen on palms, soles, and the face. The rash can sometimes be induced by heat (hot shower or towel) or by rubbing the skin (Koebner phenomenon). Arthritis can initially be mild, transient, and oligoarticular and may evolve to severe, destructive and symmetric polyarticular forms 18. Commonly involved joints are the knees, wrists, and ankles, although elbows, proximal interphalangeal joints, shoulders, metacarpophalangeal, metatarsophalangeal, hips, distal interphalangeal and temporomandibular joints can also be involved. Fusion of the wrist joint is characteristic of adult onset Still’s disease but is seen in only a few patients. Myalgia often gets worse with fever spikes. It may sometimes be severe and debilitating. Muscle weakness is not present, but case reports have described a slight elevation of serum creatine kinase and aldolase. Usually, electromyographic studies and muscle biopsies are either normal or may show a nonspecific inflammatory myopathy.
Patients can present with a sore throat at the time of initial evaluation and it frequently recurs during a disease flare. Examination in those cases typically reveals severe, nonsuppurative pharyngitis with negative bacterial cultures. A review of 341 cases of adult onset Still’s disease has described a sore throat in 69% of the patients 19. Symmetrical slightly tender lymphadenopathy is reported in one-third to two-thirds of patients, and splenomegaly in one-third to one-half of patients. Liver abnormalities include hepatomegaly ranging from 12 to 45 percent 15 and more commonly modest elevations of serum hepatic transaminases and alkaline phosphatase. About 30 to 40 percent of patients with adult onset Still’s disease have pericarditis, pleural effusions, and transient pulmonary infiltrates 15. Macrophage activation syndrome occurs in a small minority of patients but it may be underdiagnosed as suggested by retrospective studies in which macrophage activation syndrome occurred in six out of 50 patients and 21 out of 109 patients 20. Abdominal pain occurs in 1 to 48 percent of the patients. Nausea, anorexia, and weight loss can also be seen.
Adult-onset Still’s disease is a very uncommon disease. Its annual incidence has been estimated to be 0.1 to 0.4 cases per 100,000 people in Europe 21, 22. Females are affected slightly more than males. It has bimodal age distribution, the first peak between the ages of fifteen to twenty-five and the second between thirty-six to forty-six. However, about three-quarters of the patients report the onset of disease between sixteen and thirty-five years of age 23.
Diagnosis is based on review of symptoms, medical history, physical examination and possibly laboratory tests. There is no single test that can diagnose adult Still’s disease. Instead, blood tests are used to rule out other diseases with similar symptoms. Other tests, such as X-rays, may be done to check for joint inflammation or damage. Even though there is no specific diagnostic test, a serum ferritin level more than 1000ng/ml is common in adult onset Still’s disease.
Doctors use several drugs to treat adult Still’s disease. Over-the-counter or prescription nonsteroidal anti-inflammatory drugs (NSAIDs) help to reduce mild pain and inflammation. Corticosteroids, such as prednisone, are needed if the disease is severe or doesn’t respond to prescription NSAIDs. Disease-modifying drugs (DMARDs), such as methotrexate, and biologics, are needed in more severe cases or if the arthritis becomes chronic. It may be necessary to take more than one medication at a time to control symptoms.
According to the German Society of Rheumatology treatment for adult-onset Still’s disease (AOSD) guideline after determining adult-onset Still’s disease activity, treatment is usually commenced with glucocorticoids (corticosteroids) 4. And in the case of higher disease activity, methotrexate or calcineurin inhibitors (a class of drugs that suppress the immune system by binding to calcineurin, a protein that contributes to inflammation). Furthermore, anakinra, canakinumab, or tocilizumab, are introduced as glucocorticoid-sparing agents 4. In adult-onset Still’s disease cases who are not responsive to methotrexate or calcineurin inhibitors, anakinra, canakinumab, or tocilizumab should subsequently be used even in cases of lower disease activity states 4. Nonsteroidal anti-inflammatory drugs (NSAIDs) can be used temporarily for symptom control. Anakinra and canakinumab can be used as a first-line option in case of severe disease activity 4.
With adult Still’s disease, the medications may need to be taken even after symptoms go away. This is called maintenance therapy. It’s important to keep inflammation under control to prevent damage to joints and organs.
Figure 1. Adult onset Still’s disease rash
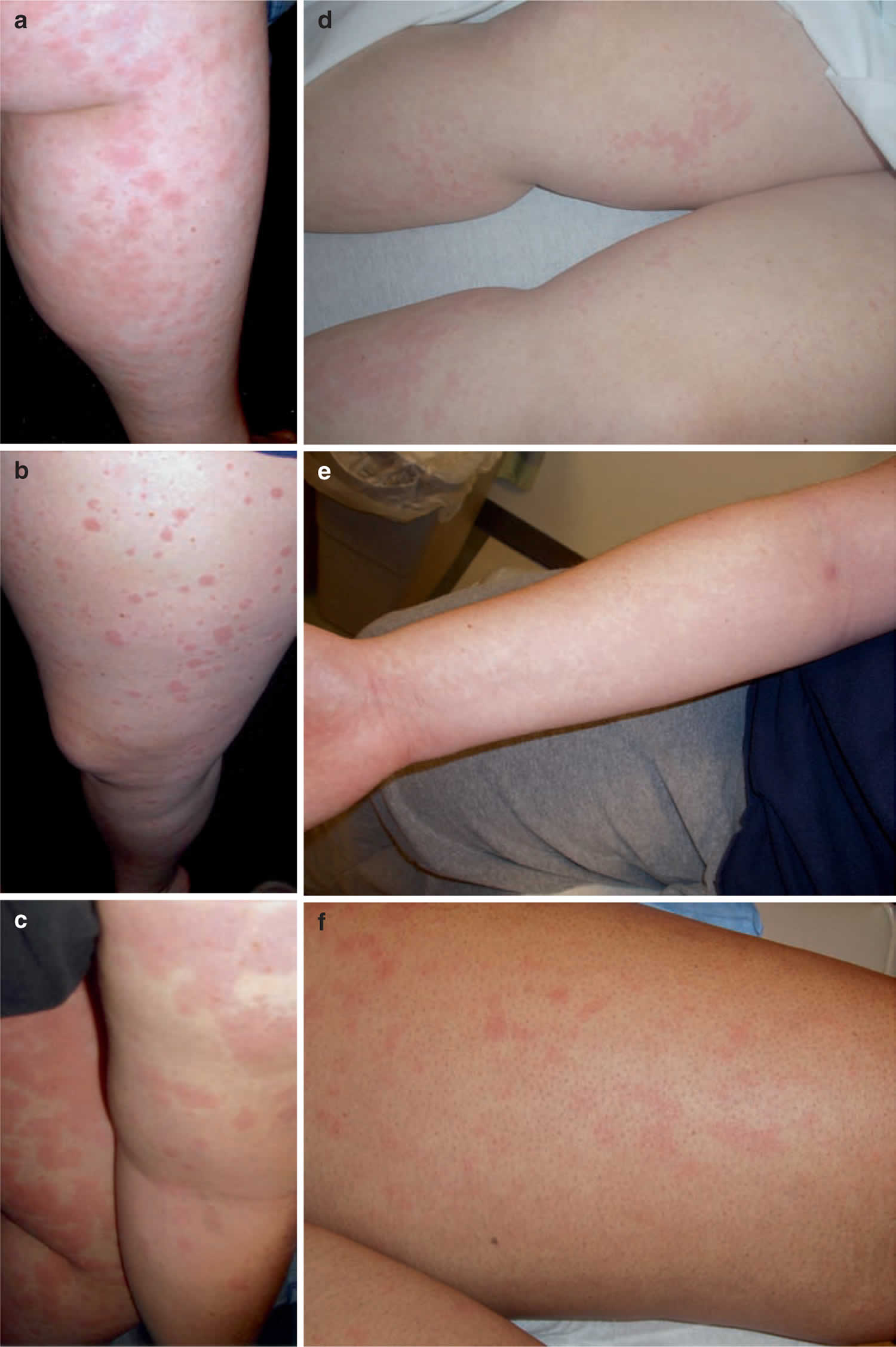
Figure 2. Clinical course of adult onset Still’s disease

Still’s disease cause
The cause of adult onset Still’s disease is unknown. Research suggests that it may be triggered by an infection. The hypothesis remains that adult onset Still’s disease is a reactive syndrome in which various infectious agents may act as triggers in a genetically predisposed host. Both genetic factors and a variety of viruses, bacteria and other infectious factors have been suggested as significant contributors to the development of adult-onset Still disease (AOSD) 24, 25. A study in Nijmegen, Netherlands evaluated for potential infectious agent through broad serological evaluation in five patients with AOSD. Two of the cases were thought to be secondary to rubella reinfection (based upon high IgG titres) while the disease in a third patient was attributed to Echovirus 7 as supported by positive culture from a throat swab as well as four-fold antibody titer increase 26. Another study evaluated 19 children with chronic rheumatic disease and isolated rubella virus from the lymphoreticular cells of seven of these patients, including one with Still’s Disease 27. Other implicated viruses include cytomegalovirus (CMV), Epstein-Barr virus (EBV), parainfluenza, parvovirus B19, human immunodeficiency virus, hepatitis A, B, and C virus, adenovirus, human herpes virus 6 and coxsackie B virus. Bacterial infectious agents to include Yersinia enterocolitica, Chlamydia trachomatis, Chlamydia pneumonia, Campylobacter jejuni, and Mycoplasma pneumoniae have also been implicated 28, 29, 30, 31.
There is uncertainty regarding the presence of same etiopathogenic factors among all the adult onset Still’s disease patients. A French study of 62 patients showed the association of adult onset Still’s disease with human leukocyte antigen (HLA) subtypes (B17, B18, B35, and DR2) 32. Specific case reports have highlighted instances of adult-onset Still disease (AOSD) occurring in twins 33.
Still’s disease pathophysiology
There are two immune dysregulations described in the pathogenesis of adult onset Still’s disease: innate and adaptive immunity 1.
Immunopathology in innate immunity:
- Activation of neutrophils and macrophages
- Increased CXCL8 levels:
- Not correlated with disease activity
- Recruits neutrophils to the inflammation site
- Associated with the persistence of chronic articular adult onset Still’s disease
- Elevated macrophage activation markers:
- Macrophage-colony stimulating factor (M-CSF)
- Calprotectin
- Intracellular adhesion molecule-1 (ICAM-1)
- Migration inhibitory factor (MIF)
- Interferon-gamma (INF-γ)
- Elevated cytokines:
- Interleukin 1β (IL-1β)
- IL-6 in skin rash specimens and serum correlating with disease activity
- IL-18 in serum, synovial fluid, liver, and lymph nodes
- Tumor necrosis factor-α (TNF) in sera and tissues
- Overexpression of toll-like receptor (TLR) 7-MyD88 pathway in dendritic cells:
- Correlates with disease activity and treatment
- Increased CXCL8 levels:
Immunopathology in adaptive immunity:
- Increased Th1 cellular immune response
- Predominance of T cells produced by IL-4T over IFN-γ in serum, skin, and synovium compared to healthy controls 34
- Increased α-soluble receptor of IL-2 (CD25)
Furthermore, the role of Th-17 responses is emerging in the pathogenesis of adult onset Still’s disease. Elevated levels of Th-17-related cytokines, including IL-1,6,17,18,21, and 23, contribute to the complex immunopathology associated with adult onset Still’s disease.
Still’s disease symptoms
The symptoms, progression and severity of adult onset Still’s disease are highly variable from one person to another. Adult onset Still’s disease symptoms usually begin with a high fever that spikes once or twice a day and a salmon-pink rash on the trunk, arms or legs. Other symptoms include sore throat and swollen lymph nodes in the neck.
Almost all people with Still’s disease will have fever, joint pain, sore throat, and a rash.
- Joint pain, warmth, and swelling are common. Most often, several joints are involved at the same time. Often, people with the condition have morning stiffness of joints that lasts for several hours.
- The fever comes on quickly once per day, most commonly in the afternoon or evening.
- The skin rash is often salmon-pink colored and comes and goes with the fever.
Additional symptoms include:
- Abdominal pain and swelling
- Pain when taking a deep breath (pleurisy)
- Sore throat
- Swollen lymph nodes (glands)
- Weight loss
The spleen or liver may become swollen. Lung and heart inflammation may also occur.
A few weeks after these initial symptoms, joints and muscles begin aching. These aches last at least two weeks. The most commonly affected joints are the knee and wrist. The ankles, shoulders, elbows and finger joints may also be involved.
Some individuals may only develop one random episode that responds to treatment and resolves within one year. In some cases, a new episode does not occur or does not occur until many years later. Other individuals may develop chronic disease, in which episodes come and go, often years apart and with no symptoms in-between episodes. Still other individuals may experience frequent episodes that occur every several weeks or months. Individuals with chronic adult onset Still’s disease may have a form predominantly characterized by fever or a form predominantly characterized by joint disease (chronic arthritis). Chronic adult onset Still’s disease can potentially cause long-term, severe and disabling complications.
Most individuals with adult onset Still’s disease develop some combination of the symptoms normally associated with systemic inflammatory disease. Such symptoms include a spiking fever greater than 102.2 degrees Fahrenheit (39 degrees Celsius), joint pain (arthralgia) and inflammation (arthritis), muscle pain (myalgia), and a skin rash.
In some cases, fevers are a daily occurrence and usually peak or spike in the late afternoon or early evening. In rare cases, some individuals develop two spiking fevers in one day. The rash is pink or salmon colored, and usually develops during a fever episode. The chest and thighs are most often affected by the rash. The arms, legs and face are affected less often. The rash may or may not be itchy and tends to disappear quickly (evanescent).
Affected joints may become swollen, stiff and inflamed and may persist for a couple of weeks. The knees, wrists and ankles are most commonly affected. Muscle and joint pain can be intense and is often worse during a fever episode. If adult onset Still’s disease goes untreated, chronic inflammation of the joints can potentially result in deterioration and destruction of the affected joints.
Additional findings may occur in some cases including a sore throat, enlargement of the spleen (splenomegaly), enlargement of the liver (hepatomegaly), and enlargement of the lymph nodes (lymphadenopathy).
In some cases, the thin, sac-like membrane that surrounds the heart (pericardium) or the heart muscle (myocardium) may become inflamed (pericarditis or myocarditis). The membrane lining the chest cavity may also become inflamed and may cause fluid to accumulate in the lungs (pleural effusion). Heart and lung involvement can cause difficulty breathing and chest pain, but in most cases it is usually not severe enough to be readily apparent and is often only detected during a physical examination by a physician.
Still’s disease complications
Adult onset Still’s disease complications include macrophage activation syndrome (MAS), amyloidosis, disseminated intravascular coagulopathy (DIC), pulmonary arterial hypertension (PAH), thrombotic thrombocytopenic purpura (TTP), and diffuse alveolar hemorrhage. Timely recognizing and managing these complications is crucial in optimizing patient outcomes and minimizing potential long-term complications.
The most severe complication of adult onset Still’s disease is macrophage activation syndrome (MAS). The prevalence ranges between 10 and 15% and is associated with a high mortality rate 35. Infections or medications, in combination with uncontrolled and prolonged inflammation in patients with a genetic predisposition, may cause this potentially fatal condition 36, 37. Macrophage activation syndrome (MAS) can occur either at the time of diagnosis or later on. Specific predictive or diagnostic factors are lacking 38. High fever, hepatosplenomegaly, cytopenias, coagulopathy, extreme hyperferritinemia, and hemophagocytosis on bone marrow aspirates are the most common symptoms of MAS.
Still’s disease diagnosis
The diagnosis of adult onset Still’s disease is difficult because there are no specific tests or distinguishing laboratory (histopathologic) findings that clearly differentiate the disorder from similar disorders. Instead, blood tests are used to rule out other diseases with similar symptoms. Other tests, such as X-rays, may be done to check for joint inflammation or damage.
Adult onset Still’s disease diagnosis is usually made based upon a thorough clinical evaluation, a detailed patient history, identification of characteristic findings, and the exclusion of other possible disorders (diagnosis of exclusion). A variety of tests may be performed to aid in a diagnosis including blood tests and x-ray studies that reveal changes in the bones or joints or enlargement of the spleen or liver. An echocardiogram, which uses sound waves to create a picture of the heart, may reveal inflammation of the pericardium or myocardium.
Blood tests may reveal characteristic changes to blood cell levels normally associated with adult onset Still’s disease. Affected individuals often have elevated levels of white blood cells and/or platelets or low levels of red blood cells. A common blood test for individuals suspected of having an inflammatory disorder is an erythrocyte sedimentation rate. Sedimentation rate measures how long it takes red blood cells (erythrocytes) to settle in a test tube over a given period. Many individuals with adult’s onset Still’s disease have an elevated sedimentation rate, which is an indication of inflammation. Another blood test commonly used is serum ferritin that is frequently disproportionally elevated in adult onset Still’s disease.
At least seven different variety of classification criteria for adult onset Still’s disease are there for use in research. The Japanese standards termed the Yamaguchi criteria 39, 40 has the highest sensitivity amongst them and is the most commonly used. The Fautrel criteria, which include ferritin and glycosylated ferritin levels as diagnostic biomarkers and do not require exclusion criteria, offer an alternative to the Yamaguchi criteria 41. In a 2018 validation study, both the Yamaguchi criteria and Fautrel criteria showed high sensitivity and specificity 42.
The lab findings discussed below are characteristic of adult onset Still’s disease, but not pathognomonic and hence their presence along with clinical manifestations will help the clinician in establishing the diagnosis after ruling out alternate causes.
Inflammatory markers, erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) are elevated in almost all the patients 43. Leukocytosis, generally more than 15,000 cells/microL with a predominance of neutrophils greater than 80%, normocytic normochromic anemia and thrombocytosis are the hematological findings. These hematological abnormalities can be severe enough to mimic primary hematologic disease. Bone marrow biopsy has been reported to show hyperplasia of granulocytic precursors and hypercellularity and hemophagocytosis in some cases. Hepatic transaminases can be elevated in 75 percent of patients and aldolase can also elevate in some due to liver inflammation.
Ferritin levels are generally higher than five times the upper limits of normal in patients with adult onset Still’s disease. Elevated ferritin suggests the presence of the disease with an 80% sensitivity and 46% specificity. If combined with a decrease in the proportion of glycosylated ferritin <20%, the specificity will rise to 93% 44.
Less than 10% of the patients have Antinuclear antibodies (ANA) and rheumatoid factor (RF) but generally only in low titer. The scarcity of these autoantibodies underscores the importance of considering additional diagnostic factors and clinical manifestations for a thorough evaluation in establishing a comprehensive diagnosis of adult onset Still’s disease.
The synovial fluid is usually inflammatory with a mean leukocyte range of 100 to 48,000 cells/microL 43. Furthermore, inflammatory markers in the synovial fluid underscore the systemic nature of adult onset Still’s disease and guide clinicians in tailoring appropriate therapeutic interventions.
Radiographs early in the disease typically are either normal or show slight joint space narrowing or periarticular osteopenia. A classic radiographic finding of adult-onset Still’s disease (AOSD) involves narrowing the wrist carpometacarpal and intercarpal joint spaces, which may progress to bone ankylosis 45, 46.
Computed tomography (CT) and F-fluorodeoxyglucose positron emission tomography (FDG-PET) can detect abnormalities, revealing lymph node enlargement, splenomegaly, pulmonary abnormalities, and hepatomegaly. These imaging modalities play a crucial role in assessing the extent of systemic involvement and contribute to a comprehensive diagnostic evaluation.
Yamaguchi criteria for classification of adult-onset Still’s disease
Major criteria are as follows:
- Fever of 39°C or higher, lasting 1 week or longer
- Arthralgia or arthritis lasting 2 weeks or longer
- Typical rash: macular or maculopapular nonpruritic salmon-pink eruption usually appearing during fever
- Granulocytic leukocytosis (≥10 x 109/l or > 10,000/mm³) including ≥80% neutrophils
Minor criteria are as follows:
- Sore throat
- Lymphadenopathy and/or splenomegaly or hepatomegaly
- Abnormal liver tests, particularly elevations in aspartate and alanine aminotransferase and lactate dehydrogenase concentrations
- Negative rheumatoid factor (RF) and antinuclear antibody (ANA)
Exclusion Criteria
- Infections
- Malignancies
- Other rheumatic diseases
Classification of adult onset Still’s disease requires 5 or more criteria including 2 or more major criteria 39. Total of five or more criteria including 2 or more significant criteria have a sensitivity of 96% and specificity of 92% to classify a patient as having adult onset Still’s disease. Any disease listed under “Exclusions” should be excluded 39.
Fautrel criteria for classification of adult-onset Still disease
Four or more major criteria are required, or 3 major and 2 minor criteria 41.
Major criteria
- Spiking fever ≥ 39°C
- Arthralgia
- Transient erythema
- Pharyngitis
- Polymorphonuclear cells ≥ 80%
- Glycosylated ferritin ≤ 20%
Minor criteria
- Maculopapular rash
- Leukocytosis ≥ 10,000/mm³
Still’s disease treatment
The goals of therapy include controlling symptoms and physical signs of inflammation as well as laboratory markers of inflammation. It also includes preventing end-organ damage and keeping the long term effects of therapy to the minimum.
The efficacy of treatment interventions for adult onset Still’s disease is derived from observational studies and clinical experience 47.
The initial therapeutic decisions are based upon the degree of disease activity and the subsequent ones based on the clinical response.
The mild disease presents with fevers, rash, arthralgias or mild arthritis. The patients with mild disease may respond to aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen, are most often used first, but most of them require at least a low dose of glucocorticoid for better control. The dose of NSAIDs is in anti-inflammatory range.
Typical nonsteroidal anti-inflammatory drugs (NSAIDs) include:
- Naproxen 500 mg twice daily
- Indomethacin 25 to 50 mg 3 times daily
- Ibuprofen 800 mg 3 times daily
The moderate adult onset Still’s disease presents with debilitating joint symptoms, high-grade fever or internal organ involvement which is not life-threatening. The patients with moderate disease severity are initially treated with glucocorticoids initiated with prednisone 0.5 to 1 mg/kg per day, depending upon disease severity. Biologic, as well as non-biologic disease-modifying antirheumatic drugs, might be needed if the steroids cannot be successfully tapered. Anakinra is favored as the initial agent in patients without joint erosions, and methotrexate is preferred in patients with prevalent joint disease.
Patients with moderate to severe adult onset Still’s disease are managed as follows:
- Anakinra is favored as the initial agent in patients without joint erosions, while methotrexate is preferred in patients with prevalent joint disease. The initial dose is 100 mg administered subcutaneously daily, with the option to increase to 100 mg twice daily if a partial response is observed within 2 weeks.
- Glucocorticoids serve as the second-line treatment after anakinra, typically at a dose of prednisone 20 to 60 mg orally. In some instances, intraarticular steroids may be considered, and a pulse dose of methylprednisolone 100 mg daily for up to three days is used in patients with macrophage activation syndrome (MAS) or those with refractory systemic or arthritic disease 48.
- Limited evidence suggests that canakinumab, rilonacept, rituximab, and abatacept may effectively treat patients refractory to other therapies.
Those with severe disease have life-threatening organ involvement like cardiac tamponade, disseminated intravascular involvement or severe liver involvement. Those with severe disease require initial therapy with pulse IV methylprednisolone 1000 mg daily for three days, and this approach has been proven effective in many case reports and series 49. Early use of biologic therapy is also recommended in severe disease. Among biologics, IL-1 inhibitor Anakinra and IL-6 inhibitor Tocilizumab are more effective than TNF inhibitors. Limited evidence suggests that Canakinumab, Rilonacept, Rituximab, and Abatacept may be effective in treating patients who are refractory to therapies as mentioned above.
Patients should undergo regular monitoring, including periodic assessments of complete blood count (CBC), blood urea nitrogen (BUN), creatinine, electrolytes, ferritin, D-dimer, alanine transaminase (ALT), and aspartate aminotransferase (AST).
Most patients with adult onset Still’s disease eventually achieve the ability to discontinue therapy. Some may experience a monocyclic form of adult onset Still’s disease. There are no established protocols for tapering and discontinuing disease-modifying antirheumatic drugs.
For patients in complete remission for at least 3 months, the recommended approach is to taper medications to discontinue all drugs gradually. Regular monitoring during tapering contributes to individualized management for each patient with adult onset Still’s disease.
Self care
Caring for the body and mind are key components of an adult Still’s disease management plan. Make positive and healthy lifestyle choices and acknowledge the physical and emotional effects of arthritis. Proper nutrition, activity, rest and following doctors’ orders are important for managing the condition and possible medication side-effects.
Still’s disease prognosis
The course of adult onset Still’s disease generally follows one of the three patterns – self-limited illness (monophasic), intermittent flares (polycyclic) or chronic Still’s disease 6, 35, 50, 51, 52, 32, 53, 54. The prognosis for adult onset Still’s disease is good with an estimated specific mortality rate of 1–3% 36. Some patients, however, experience complications. Even so, early diagnosis and prognosis assessment may help to reduce adult onset Still’s disease’s critical problems, such as macrophage activation syndrome (MAS), thrombotic thrombocytopenic purpura (TTP), respiratory distress syndrome, and diffuse alveolar hemorrhage 55. The disease’s multi-visceral involvement may significantly reduce AOSD patients’ life expectancy.
There are several predictors for the chronicity of the disease and the unfavorable outcomes. It includes the presence of an erosive polyarthritis at the time of presentation and involvement of shoulders or hips 43, 56, 57. Additionally, the need for systemic glucocorticoids for more than 2 years before routine use of biologics is also considered a poor prognostic marker 1.
- Bhargava J, Panginikkod S. Still Disease. [Updated 2024 Feb 26]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538345[↩][↩][↩]
- Kurosawa Y, Takamura S, Wakamatsu A, Kobayashi D, Narita I. A Case of Adult-Onset Still’s Disease With Hypocomplementemia. Cureus. 2024 Jan 20;16(1):e52605. doi: 10.7759/cureus.52605[↩]
- Macovei LA, Burlui A, Bratoiu I, Rezus C, Cardoneanu A, Richter P, Szalontay A, Rezus E. Adult-Onset Still’s Disease-A Complex Disease, a Challenging Treatment. Int J Mol Sci. 2022 Oct 24;23(21):12810. doi: 10.3390/ijms232112810[↩][↩]
- Vordenbäumen S, Feist E, Rech J, Fleck M, Blank N, Haas JP, Kötter I, Krusche M, Chehab G, Hoyer B, Kiltz U, Fell D, Reiners J, Weseloh C, Schneider M, Braun J. Diagnosis and treatment of adult-onset Still’s disease: a concise summary of the German society of rheumatology S2 guideline. Z Rheumatol. 2023 Feb;82(Suppl 2):81-92. English. doi: 10.1007/s00393-022-01294-2. Epub 2022 Dec 15. Erratum in: Z Rheumatol. 2023 Feb;82(Suppl 2):93-94. doi: 10.1007/s00393-023-01337-2[↩][↩][↩][↩][↩]
- Anderson CW, Shah PA, Roberts JR. Adult-Onset Still’s Disease: Is This Truly a Diagnosis of Exclusion? Hawaii J Med Public Health. 2017 Nov;76(11 Suppl 2):3-6. https://pmc.ncbi.nlm.nih.gov/articles/PMC5696586[↩]
- Feist E., Mitrovic S., Fautrel B. Mechanisms, Biomarkers and Targets for Adult-Onset Still’s Disease. Nat. Rev. Rheumatol. 2018;14:603–618. doi: 10.1038/s41584-018-0081-x[↩][↩]
- Evensen KJ, Swaak TJG, Nossent JC (2007) Increased ferritin response in adult Still’s disease: specificity and relationship to outcome. Scand J Rheumatol 36:107–110. https://doi.org/10.1080/03009740600958504[↩]
- Fautrel B, Le Moël G, Saint-Marcoux B, Taupin P, Vignes S, Rozenberg S, Koeger AC, Meyer O, Guillevin L, Piette JC, Bourgeois P. Diagnostic value of ferritin and glycosylated ferritin in adult onset Still’s disease. J Rheumatol. 2001 Feb;28(2):322-9. https://www.jrheum.org/content/28/2/322.long[↩]
- Zhang M, Wang Y, Li J, Zhou J (2020) Adult-onset Still’s disease presenting as fever of unknown origin: a single-center retrospective observational study from China. Ann Palliat Med 9:2786–2792. https://doi.org/10.21037/apm-20-268[↩]
- Vanderschueren S, Hermans F, De Munter P, Knockaert D. Adult-onset Still’s disease: still a diagnosis of exclusion. A nested case-control study in patients with fever of unknown origin. Clin Exp Rheumatol. 2012 Jul-Aug;30(4):514-9. https://www.clinexprheumatol.org/abstract.asp?a=5493[↩]
- Uppal SS, Al-Mutairi M, Hayat S et al (2007) Ten years of clinical experience with adult onset Still’s disease: is the outcome improving? Clin Rheumatol 26:1055–1060. https://doi.org/10.1007/s10067-006-0440-x[↩]
- Sun Y, Wang Z, Chi H et al (2019) Elevated serum levels of interleukin-10 in adult-onset Still’s disease are associated with disease activity. Clin Rheumatol 38:3205–3210. https://doi.org/10.1007/s10067-019-04642-x[↩]
- Ruscitti P, Cipriani P, Masedu F et al (2016) Adult-onset Still’s disease: evaluation of prognostic tools and validation of the systemic score by analysis of 100 cases from three centers. BMC Med 14:194. https://doi.org/10.1186/s12916-016-0738-8[↩]
- Reddy Munagala VV, Misra R, Agarwal V et al (2012) Adult onset Still’s disease: experience from a tertiary care rheumatology unit. Int J Rheum Dis 15:e136–141. https://doi.org/10.1111/1756-185X.12012[↩]
- Gerfaud-Valentin M, Jamilloux Y, Iwaz J, Sève P. Adult-onset Still’s disease. Autoimmun Rev. 2014 Jul;13(7):708-22.[↩][↩][↩]
- Calabro JJ, Marchesano JM. Fever associated with juvenile rheumatoid arthritis. N. Engl. J. Med. 1967 Jan 05;276(1):11-8.[↩]
- Gerfaud-Valentin M, Maucort-Boulch D, Hot A, Iwaz J, Ninet J, Durieu I, Broussolle C, Sève P. Adult-onset still disease: manifestations, treatment, outcome, and prognostic factors in 57 patients. Medicine (Baltimore). 2014 Mar;93(2):91-9.[↩]
- Elkon KB, Hughes GR, Bywaters EG, Ryan PF, Inman RD, Bowley NB, James MP, Eady RA. Adult-onset Still’s disease. Twenty-year followup and further studies of patients with active disease. Arthritis Rheum. 1982 Jun;25(6):647-54.[↩]
- Nguyen KH, Weisman MH. Severe sore throat as a presenting symptom of adult onset Still’s disease: a case series and review of the literature. J. Rheumatol. 1997 Mar;24(3):592-7.[↩]
- Bae CB, Jung JY, Kim HA, Suh CH. Reactive hemophagocytic syndrome in adult-onset Still disease: clinical features, predictive factors, and prognosis in 21 patients. Medicine (Baltimore). 2015 Jan;94(4):e451.[↩]
- Mitrovic S., Fautrel B. Clinical Phenotypes of Adult-Onset Still’s Disease: New Insights from Pathophysiology and Literature Findings. J. Clin. Med. 2021;10:2633. doi: 10.3390/jcm10122633[↩]
- Galozzi P., Bindoli S., Doria A., Sfriso P. Progress in Biological Therapies for Adult-Onset Still’s Disease. Biologics. 2022;16:21–34. doi: 10.2147/BTT.S290329[↩]
- Maruyama A., Kokuzawa A., Yamauchi Y., Kirino Y., Nagai H., Inoue Y., Ota T., Chifu Y., Inokuchi S., Koarada S., et al. Clinical Features of Elderly-Onset Adult-Onset Still’s Disease. Mod. Rheumatol. 2020;31:862–868. doi: 10.1080/14397595.2020.1829340[↩]
- Ohta A, Yamaguchi M, Tsunematsu T, Kasukawa R, Mizushima H, Kashiwagi H, Kashiwazaki S, Tanimoto K, Matsumoto Y, Akizuki M, et al. Adult Still’s disease: a multicenter survey of Japanese patients. J Rheumatol. 1990 Aug;17(8):1058-63.[↩]
- Colebunders R, Stevens WJ, Vanagt E, Snoeck J. Adult Still’s Disease Caused by Yersinia enterocolitica Infection. Arch Intern Med. 1984;144(9):1880–1882. doi:10.1001/archinte.1984.00350210210040[↩]
- Wouters JM, van der Veen J, van de Putte LB, de Rooij DJ. Adult onset Still’s disease and viral infections. Ann Rheum Dis. 1988 Sep;47(9):764–767. doi: 10.1136/ard.47.9.764[↩]
- Chantler JK, Tingle AJ, Petty RE. Persistent rubella virus infection associated with chronic arthritis in children. N Engl J Med. 1985 Oct;313(18):1117–1123. doi: 10.1056/NEJM198510313131803[↩]
- Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol. 2008 Oct;22(5):773–792. doi: 10.1016/j.berh.2008.08.006[↩]
- Pouchot J, Sampalis JS, Beaudet F, Carette S, Décary F, Salusinsky-Sternbach M, Hill RO, Gutkowski A, Harth M, Myhal D, et al. Adult Still’s disease: manifestations, disease course, and outcome in 62 patients. Medicine (Baltimore) 1991 Mar;70(2):118–136.[↩]
- Pouchot J, Ouakil H, Debin ML, Vinceneux P. Adult Still’s disease associated with acute human parvovirus B19 infection. Lancet. 1993 May;341(8855):1280–1281. doi: 10.1016/0140-6736(93)91184-n[↩]
- Perez C, Artola V. Adult Still’s disease associated with Mycoplasma pneumonia infection. Clin Infect Dis. 2001 Mar;32(6) doi: 10.1086/319342[↩]
- Pouchot J, Sampalis JS, Beaudet F, Carette S, Décary F, Salusinsky-Sternbach M, Hill RO, Gutkowski A, Harth M, Myhal D, et al. Adult Still’s disease: manifestations, disease course, and outcome in 62 patients. Medicine (Baltimore). 1991 Mar;70(2):118-36.[↩][↩]
- Kahn MF. Adult Still’s disease. Still many issues unresolved. J Rheumatol. 1996 Dec;23(12):2015-6.[↩]
- Chen DY, Lan JL, Lin FJ, Hsieh TY, Wen MC. Predominance of Th1 cytokine in peripheral blood and pathological tissues of patients with active untreated adult onset Still’s disease. Ann Rheum Dis. 2004 Oct;63(10):1300-6. doi: 10.1136/ard.2003.013680[↩]
- Tomaras S., Goetzke C., Kallinich T., Feist E. Adult-Onset Still’s Disease: Clinical Aspects and Therapeutic Approach. J. Clin. Med. 2021;10:733. doi: 10.3390/jcm10040733[↩][↩]
- Mehta B., Ibrahim S., Briggs W., Efthimiou P. Racial/Ethnic Variations in Morbidity and Mortality in Adult Onset Still’s Disease: An Analysis of National Dataset. Semin. Arthritis Rheum. 2019;49:469–473. doi: 10.1016/j.semarthrit.2019.04.004[↩][↩]
- Crayne C., Albeituni S., Nichols K., Cron R. The Immunology of Macrophage Activation Syndrome. Front. Immunol. 2019;10:119. doi: 10.3389/fimmu.2019.00119[↩]
- Machaczka M., Klimkowska M. Bone Marrow Assessment in the Diagnosis of Acquired Hemophagocytic Lymphohistiocytosis in Adults. Am. J. Clin. Pathol. 2015;143:308–309. doi: 10.1309/AJCPUK8TLI2MLYOQ[↩]
- Yamaguchi M, Ohta A, Tsunematsu T, Kasukawa R, Mizushima Y, Kashiwagi H, Kashiwazaki S, Tanimoto K, Matsumoto Y, Ota T, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992 Mar;19(3):424-30.[↩][↩][↩]
- Fautrel B, Zing E, Golmard JL, Le Moel G, Bissery A, Rioux C, Rozenberg S, Piette JC, Bourgeois P. Proposal for a new set of classification criteria for adult-onset still disease. Medicine (Baltimore). 2002 May;81(3):194-200.[↩]
- Validation of the Fautrel classification criteria for adult-onset Still’s disease. Lebrun D, Mestrallet S, Dehoux M, et al. Semin Arthritis Rheum. 2018;47:578–585. doi: 10.1016/j.semarthrit.2017.07.005[↩][↩]
- Mechanisms, biomarkers and targets for adult-onset Still’s disease. Feist E, Mitrovic S, Fautrel B. Nat Rev Rheumatol. 2018;14:603–618. doi: 10.1038/s41584-018-0081-x[↩]
- Pouchot J, Sampalis JS, Beaudet F, Carette S, Décary F, Salusinsky-Sternbach M, Hill RO, Gutkowski A, Harth M, Myhal D. Adult Still’s disease: manifestations, disease course, and outcome in 62 patients. Medicine (Baltimore). 1991 Mar;70(2):118-36.[↩][↩][↩]
- Van Reeth C, Le Moel G, Lasne Y, Revenant MC, Agneray J, Kahn MF, Bourgeois P. Serum ferritin and isoferritins are tools for diagnosis of active adult Still’s disease. J Rheumatol. 1994 May;21(5):890-5.[↩]
- Medsger TA Jr, Christy WC. Carpal arthritis with ankylosis in late onset Still’s disease. Arthritis Rheum. 1976 Mar-Apr;19(2):232-42. doi: 10.1002/art.1780190216[↩]
- Björkengren AG, Pathria MN, Sartoris DJ, Terkeltaub R, Esdaile JM, Weisman M, Resnick D. Carpal alterations in adult-onset Still disease, juvenile chronic arthritis, and adult-onset rheumatoid arthritis: comparative study. Radiology. 1987 Nov;165(2):545-8. doi: 10.1148/radiology.165.2.3659381[↩]
- Pouchot J, Arlet JB. Biological treatment in adult-onset Still’s disease. Best Pract Res Clin Rheumatol. 2012 Aug;26(4):477-87.[↩]
- Khraishi M, Fam AG. Treatment of fulminant adult Still’s disease with intravenous pulse methylprednisolone therapy. J Rheumatol. 1991 Jul;18(7):1088-90.[↩]
- Franchini S, Dagna L, Salvo F, Aiello P, Baldissera E, Sabbadini MG. Efficacy of traditional and biologic agents in different clinical phenotypes of adult-onset Still’s disease. Arthritis Rheum. 2010 Aug;62(8):2530-5.[↩]
- Bagnari V., Colina M., Ciancio G., Govoni M., Trotta F. Adult-Onset Still’s Disease. Rheumatol. Int. 2009;30:855–862. doi: 10.1007/s00296-009-1291-y[↩]
- Efthimiou P., Kontzias A., Ward C., Ogden N. Adult-Onset Still’s Disease: Can Recent Advances in Our understanding of Its Pathogenesis Lead to Targeted Therapy? Nat. Rev. Rheumatol. 2007;3:328–335. doi: 10.1038/ncprheum0510[↩]
- Pouchot J., Sampalis J., Beaudet F., Carette S., Decary F., Salusinky-Sternbach M., Hill R., Gutkowski A., Harth M., Myhal D., et al. Adult Still’s Disease. Medicine. 1991;70:118–136. doi: 10.1097/00005792-199103000-00004[↩]
- Kontzias A, Efthimiou P. Adult-onset Still’s disease: pathogenesis, clinical manifestations and therapeutic advances. Drugs. 2008;68(3):319-37. doi: 10.2165/00003495-200868030-00005. Erratum in: Drugs. 2011 Oct 1;71(14):1820[↩]
- Gerfaud-Valentin M, Maucort-Boulch D, Hot A, Iwaz J, Ninet J, Durieu I, Broussolle C, Sève P. Adult-onset still disease: manifestations, treatment, outcome, and prognostic factors in 57 patients. Medicine (Baltimore). 2014 Mar;93(2):91-99. doi: 10.1097/MD.0000000000000021[↩]
- Kaneko Y. Interluekin-6 Inhibitors for the Treatment of Adult-Onset Still’s Disease. Mod. Rheumatol. 2021;32:12–15. doi: 10.1093/mr/roab004[↩]
- Calabro JJ, Marchesano JM. Fever associated with juvenile rheumatoid arthritis. N Engl J Med. 1967 Jan 5;276(1):11-8. doi: 10.1056/NEJM196701052760102[↩]
- Wouters JM, van der Veen J, van de Putte LB, de Rooij DJ. Adult onset Still’s disease and viral infections. Ann Rheum Dis. 1988 Sep;47(9):764-7. https://pmc.ncbi.nlm.nih.gov/articles/instance/1003594/pdf/annrheumd00419-0060.pdf[↩]