Bethlem myopathy is predominantly an autosomal dominant myopathy, classified as a congenital form of limb-girdle muscular dystrophy.[2] There are two types of Bethlem myopathy, based on which type of collagen is affected.[3]
| Bethlem myopathy | |
|---|---|
| Other names | Muscular dystrophy, limb-girdle, autosomal dominant 5, (LGMDD5); Muscular dystrophy, limb-girdle, autosomal recessive 22, (LGMDR22); Ehlers–Danlos syndrome, myopathic type (EDSMYP) |
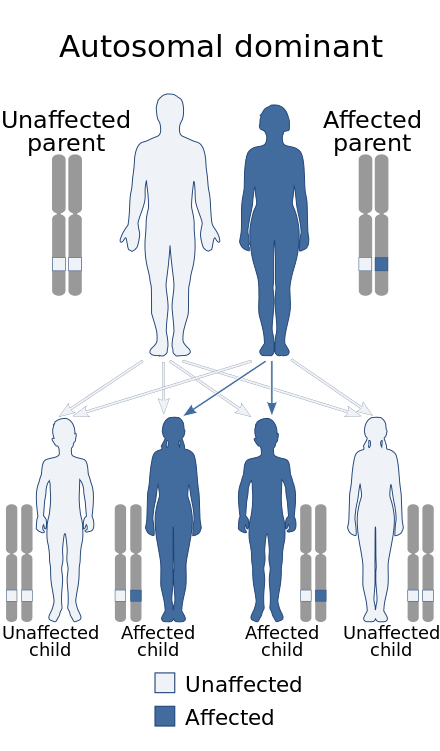 | |
| Bethlem myopathy has an autosomal dominant pattern of inheritance (autosomal recessive form exists as well[1]). | |
Bethlem myopathy 1 (BTHLM1) is caused by a mutation in one of the three genes coding for type VI collagen.[4][3] These include COL6A1, COL6A2, and COL6A3.[5][3] It is typically autosomal dominant, though uncommonly can be autosomal recessive.[3]
Bethlem myopathy 2 (BTHLM2), formerly known as myopathic-type Ehlers–Danlos syndrome, is caused by a mutation on the COL12A1 gene coding for type XII collagen.[3] It is autosomal dominant.[3]
In 2017, an international workshop proposed a redefined criteria and naming system for limb-girdle muscular dystrophies. Bethlem myopathy 1 (collagen VI) was included into the proposed list and renamed LGMDD5 for autosomal dominant mutations and LGMDR22 for recessive mutations. Bethlem myopathy 2 (collagen XII) was not addressed.[2]
Gowers's sign, toe walking, multiple contractures of the joints (especially the fingers: 'Bethlem sign'), skin abnormalities, and muscle weakness (proximal more than distal) are typical signs and symptoms of the disease. Initially, in early childhood, there may also be joint laxity. There is no cardiac involvement in either Bethlem myopathy 1 or 2, which helps to differentiate it from Emery–Dreifuss muscular dystrophy.[6] Currently there is no cure for the disease and symptomatic treatment is used to relieve symptoms and improve quality of life.[7]
Bethlem myopathy may be diagnosed based on clinical examinations and laboratory tests may be recommended. Genetic testing for known pathological variants is preferred. In the case of a VUS, testing of dermal fibroblast culture is used for an accurate diagnosis.[6]
Bethlem myopathy 1 is a rare disease, affecting about 1 in 200,000 people.[8] Bethlem myopathy 2 is an ultra-rare disease, affecting less than 1 in 1,000,000 people.[9]
The condition was described by J. Bethlem and G. K. van Wijngaarden in 1976.[10]
Signs and symptoms
editBethlem myopathy is a slowly progressive muscle disease characterized predominantly by contractures, rigidity of the spine, skin abnormalities and proximal muscle weakness.[5][11] Symptoms may present as early as infancy, with typical contractures and hyperlaxity of joints; however, in some patients, symptoms may go unnoticed until adolescence or adulthood.[11] Serum creatine kinase (CK) is usually normal to mildly elevated (<5×).[11]
Early on, there may be distal laxity (hypermobility), but all of those with Bethlem myopathy eventually develop multiple joint contractures: long finger flexors, wrists, elbows, hips, knees and ankles.[5][11] There may also be club foot, scoliosis or rigid spine.[5][11] Skin abnormalities are common, including keloid formation, ‘cigarette paper scarring’ (atrophic scarring), velvety soft skin, and follicular hyperkeratosis.[11][6]
'Bethlem sign' is the typical sign in Bethlem myopathy patients demonstrating long finger flexor contractures. With palms facing each other and with elbows raised, patients try, but fail, to make full contact of one hand against the other (in what looks like the gesture of hands during prayer).[12]
Bethlem myopathy 1
edit(Collagen VI genes)
See Bethlem myopathy 1 Clinical synopsis on OMIM: 158810
In Bethlem myopathy 1, in the calf, one of the first signs is often a 'rim' of fatty infiltration between the soleus and gastrocnemius muscles.[12][13][14] Although there is fatty infiltration, the calf muscles do not appear pseudohypertrophic, in fact they may appear slender.[15][12][16][17][18] In the thighs, there is also significant fatty infiltration of the vasti muscles, with a rim of fatty infiltration on the periphery of the muscles, while the center is more or less spared (characteristic "outside-in" pattern).[12][19] This "outside-in" pattern distinguishes it from other myopathies known to have contractures, such as Emery-Dreifuss muscular dystrophy.[12]
The exception is the rectus femoris muscle of the thigh, where fatty infiltration occurs in the center of the muscle, but spares the periphery. This unusual pattern is described as a "central cloud" and is also a distinguishing feature, as it is not seen in the rectus femoris of LMNA-related Emery-Dreifuss myopathy.[12]
Bethlem myopathy 1 may also include neonatal-onset torticollis (neck contracture) and hypotonia ("floppy baby"), delayed motor mile stones, with respiratory difficulties potentially occurring later in life.[20][12] Contractures presenting in infancy may resolve by age 2 years, but reoccur as the disease progresses, typically by late of the first decade or early teens.[12]
Bethlem myopathy 2
edit(Collagen XII gene)
See Bethlem myopathy 2 Clinical synopsis on OMIM: 616471
In Bethlem myopathy 2, there is phenotypic variability. In one family, the only notable finding on T1-weighted MR images (used to detect fatty infiltration) was atrophy of the rectus femoris muscles of the thigh, with the degree of atrophy matching the severity of the disease, but no fatty infiltration.[14][11] In another family, only the more severely affected older patient showed significant abnormality, by having symmetrical fatty atrophy of the femoral quadriceps of the thigh, the adductor and medial gastrocnemius muscles of the calf; as well as asymmetrical fatty atrophy of the adductor longus of the thigh.[11] No muscle hypertrophy was reported and the muscles of the patients without fatty atrophy appeared normal.[11]
Bethlem myopathy 2 also differs by including the possibility of scapula winging, pectus excavatum, stooped posture, kyphosis (hunchback), micrognathia, retrognathia, and a high-arched palate.[14] Childhood muscle weakness improves in teen years, but muscle weakness returns by the third decade of life.[11]
Diagnosis
editThe disease may be diagnosed based on a clinical examination, which identifies signs and symptoms generally associated with the people who have the condition. Genetic testing for known pathological variants is preferred, by testing of the COL6A1, COL6A2, COL6A3 and COL12A1 genes.[7][3] In the case of a VUS, testing of dermal fibroblast culture is used for an accurate diagnosis.[6]
Additional laboratory tests may be performed before genetic testing, such as creatine kinase (CK) blood test, MRI of the muscles, and electromyography (EMG).
Phenotypes of overlap between Ullrich congenital muscular dystrophy (UCMD) and Bethlem can be assumed. In the differential diagnosis of UCMD, even in patients without finger contractures, Bethlem myopathy could be considered.[21]
Differential diagnosis
editUllrich congenital muscular dystrophy (UCMD) involves mutations on the same genes as Bethlem myopathy, but has a more severe presentation, with the ability to walk (ambulation) typically being lost between the ages of 5–15 years.[12] Autosomal recessive myosclerosis myopathy is allelic to the COL6A2 gene, it includes multiple contractures of the joints with slender muscles which are infiltrated by connective tissue and fibrosis, giving them a firm, "woody" feel upon palpitation.[22][23]
The symptoms of Bethlem myopathy may overlap with other conditions including Emery–Dreifuss muscular dystrophy, congenital muscular dystrophies, limb girdle muscular dystrophies, FHL1-related myopathies (X-linked myopathy with postural muscle atrophy, reducing body myopathy, and scapuloperoneal myopathy), and some forms of Ehlers–Danlos syndrome.[11] Tubular aggregate myopathy (TAM1 & TAM2) includes, among other symptoms, contractures, muscle weakness, and fatty atrophy of muscle.[24][25][26]
Typical to Bethlem myopathy 1 and 2 are the presence of multiple contractures.[11][5] A contracture can be caused by a variety of reasons, from disease to lifestyle (see Muscle contractures). If the patient lacks multiple contractures, as well as lacks other common symptoms of Bethlem myopathy, and in addition has muscular symptoms which are not known to be associated with Bethlem myopathy such as muscle hypertrophy, exercise-induced (dynamic) symptoms rather than fixed muscle weakness (static) symptoms, or cardiac involvement such as arrhythmia, then other myopathies should be considered.
Treatment
editCurrently there is no cure for the disease. Symptomatic treatment, which aims to relieve symptoms and improve quality of life is the main treatment method of Bethlem myopathy. It is believed that physical therapy, stretching exercises, orthoses such as braces and splints, and mobility aids like a walker or wheelchair are beneficial to patient's condition.[7]
Surgical options could be considered in rare instances, in order to help with joint contractures or scoliosis.[7] Contractures of the legs can be alleviated with heel-cord surgery followed by bracing and regular physical therapy. Repeated surgeries to lengthen the heel cords may be needed as the child grows to adulthood.[4]
Epidemiology
editAccording to a Japanese study from 2007, Bethlem myopathy 1 affects about 1 in 200,000 people.[8] A 2009 study, concerning the prevalence of genetic muscle disease in Northern England, estimated the prevalence of Bethlem myopathy 1 to be at 0.77:100,000.[27] Together with Ullrich congenital muscular dystrophy 1, Bethlem myopathy 1 is believed to be underdiagnosed. Both conditions have been described in individuals from a variety of ethnic backgrounds.[28] Bethlem myopathy 2 affects less than 1 in 1,000,000 people.[9]
References
edit- ^ RESERVED, INSERM US14 -- ALL RIGHTS. "Orphanet: Bethlem myopathy". www.orpha.net. Retrieved 23 December 2017.
{{cite web}}: CS1 maint: numeric names: authors list (link) - ^ a b Straub, Volker; Murphy, Alexander; Udd, Bjarne; LGMD workshop study group (August 2018). "229th ENMC international workshop: Limb girdle muscular dystrophies - Nomenclature and reformed classification Naarden, the Netherlands, 17-19 March 2017". Neuromuscular Disorders. 28 (8): 702–710. doi:10.1016/j.nmd.2018.05.007. ISSN 1873-2364. PMID 30055862.
- ^ a b c d e f g "Phenotypic Series - PS158810 Bethlem myopathy - PS158810 - 4 Entries". OMIM - Online Mendelian Inheritance in Man.
- ^ a b Jobsis GJ, Boers JM, Barth PG, de Visser M (1999). "Bethlem myopathy: a slowly-progressive congenital muscular dystrophy with contractures". Brain. 122 (4): 649–655. doi:10.1093/brain/122.4.649. PMID 10219778.
- ^ a b c d e Lampe AK, Bushby KM (September 2005). "Collagen VI related muscle disorders". J. Med. Genet. 42 (9): 673–85. doi:10.1136/jmg.2002.002311. PMC 1736127. PMID 16141002.
- ^ a b c d Bertini, Enrico; D'Amico, Adele; Gualandi, Francesca; Petrini, Stefania (December 2011). "Congenital muscular dystrophies: a brief review". Seminars in Pediatric Neurology. 18 (4): 277–288. doi:10.1016/j.spen.2011.10.010. ISSN 1558-0776. PMC 3332154. PMID 22172424.
- ^ a b c d "Bethlem myopathy | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2020-10-19.
- ^ a b Okada, M.; Kawahara, G.; Noguchi, S.; Sugie, K.; Murayama, K.; Nonaka, I.; Hayashi, Y. K.; Nishino, I. (2007-09-04). "Primary collagen VI deficiency is the second most common congenital muscular dystrophy in Japan". Neurology. 69 (10): 1035–1042. doi:10.1212/01.wnl.0000271387.10404.4e. ISSN 1526-632X. PMID 17785673.
- ^ a b "Myopathic EDS (mEDS)". The Ehlers Danlos Society. Retrieved 2023-05-01.
- ^ "Entry - #158810 - BETHLEM MYOPATHY 1; BTHLM1 - OMIM". Online Mendelian Inheritance in Man. Retrieved 2023-09-15.
- ^ a b c d e f g h i j k l Hicks, D.; Farsani, G. T.; Laval, S.; Collins, J.; Sarkozy, A.; Martoni, E.; Shah, A.; Zou, Y.; Koch, M.; Bonnemann, C. G.; Roberts, M.; Lochmuller, H.; Bushby, K.; Straub, V. (2014-05-01). "Mutations in the collagen XII gene define a new form of extracellular matrix-related myopathy". Human Molecular Genetics. 23 (9): 2353–2363. doi:10.1093/hmg/ddt637. ISSN 0964-6906. PMID 24334769.
- ^ a b c d e f g h i Bönnemann, Carsten G. (2011-06-21). "The collagen VI-related myopathies: muscle meets its matrix". Nature Reviews. Neurology. 7 (7): 379–390. doi:10.1038/nrneurol.2011.81. ISSN 1759-4758. PMC 5210181. PMID 21691338.
- ^ Morrow, Jasper M.; Pitceathly, Robert D. S.; Quinlivan, Ros M.; Yousry, Tarek A. (2013-04-16). "Muscle MRI in Bethlem myopathy". BMJ Case Reports. 2013: bcr2013008596. doi:10.1136/bcr-2013-008596. ISSN 1757-790X. PMC 3645630. PMID 23595177.
- ^ a b c "# 616471 BETHLEM MYOPATHY 2; BTHLM2 Alternative titles; symbols EHLERS-DANLOS SYNDROME, MYOPATHIC TYPE; EDSMYP EDS, MYOPATHIC TYPE". OMIM - Online Mendelian Inheritance in Man.
- ^ Nalini, A; Gayathri, N (2010). "Bethlem myopathy: A study of two families". Neurology India. 58 (4): 665–666. doi:10.4103/0028-3886.68684. ISSN 0028-3886. PMID 20739820.
- ^ Suh, B.; Choi, Young-Chul; Kim, S. M.; Choi, Byung-Ok; Shim, D.; Lee, D. H.; Sunwoo, I. (2006). "A Family of Bethlem Myopathy". Journal of the Korean Neurological Association. S2CID 74251729.
- ^ Souza, Paulo Victor Sgobbi de; Bortholin, Thiago; Pinheiro, Jhonatan Rafael Siqueira; Naylor, Fernando George Monteiro; Pinto, Wladimir Bocca Vieira de Rezende; Oliveira, Acary Souza Bulle (2017-10-01). "Collagen type VI-related myopathy". Practical Neurology. 17 (5): 406–407. doi:10.1136/practneurol-2017-001661. ISSN 1474-7758. PMID 28578317. S2CID 755149.
- ^ Telles, Juliana Aparecida Rhein; Voos, Mariana Calil; Anequini, Isabella Pessa; Favero, Francis Meire; Silva, Thiago Henrique; Caromano, Fátima Aparecida (June 2018). "Genetic and functional differences between Bethlem miopathyand Ullrich congenital muscular dystrophy - case studies". Cadernos de Pós-Graduação em Distúrbios do Desenvolvimento. 18 (1): 148–163. doi:10.5935/cadernosdisturbios.v18n1p148-163. ISSN 1519-0307.
- ^ Mercuri, Eugenio; Lampe, Anne; Allsop, Joanna; Knight, Ravi; Pane, Marika; Kinali, Maria; Bonnemann, Carsten; Flanigan, Kevin; Lapini, Ilaria; Bushby, Kate; Pepe, Guglielmina; Muntoni, Francesco (April 2005). "Muscle MRI in Ullrich congenital muscular dystrophy and Bethlem myopathy". Neuromuscular Disorders. 15 (4): 303–310. doi:10.1016/j.nmd.2005.01.004. ISSN 0960-8966. PMID 15792870. S2CID 25600853.
- ^ "BETHLEM MYOPATHY 1; BTHLM1". www.omim.org. Retrieved 2023-09-30.
- ^ Reed, Umbertina Conti; Ferreira, Lucio Gobbo; Liu, Enna Cristina; Resende, Maria Bernadete Dutra; Carvalho, Mary Souza; Marie, Suely Kazue; Scaff, Milberto (September 2005). "Ullrich congenital muscular dystrophy and bethlem myopathy: clinical and genetic heterogeneity". Arquivos de Neuro-Psiquiatria. 63 (3B): 785–790. doi:10.1590/S0004-282X2005000500013. ISSN 0004-282X. PMID 16258657.
- ^ Merlini, L.; Martoni, E.; Grumati, P.; Sabatelli, P.; Squarzoni, S.; Urciuolo, A.; Ferlini, A.; Gualandi, F.; Bonaldo, P. (2008-10-14). "Autosomal recessive myosclerosis myopathy is a collagen VI disorder". Neurology. 71 (16): 1245–1253. doi:10.1212/01.wnl.0000327611.01687.5e. ISSN 1526-632X. PMID 18852439. S2CID 21554344.
- ^ "MYOSCLEROSIS, AUTOSOMAL RECESSIVE". www.omim.org. Retrieved 2023-10-13.
- ^ "MYOPATHY, TUBULAR AGGREGATE, 1; TAM1". www.omim.org. Retrieved 2023-11-11.
- ^ "MYOPATHY, TUBULAR AGGREGATE, 2; TAM2". www.omim.org. Retrieved 2023-11-11.
- ^ "Tubular aggregate myopathy - About the Disease - Genetic and Rare Diseases Information Center". rarediseases.info.nih.gov. Retrieved 2023-11-11.
- ^ Norwood, Fiona L. M.; Harling, Chris; Chinnery, Patrick F.; Eagle, Michelle; Bushby, Kate; Straub, Volker (2009). "Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population". Brain: A Journal of Neurology. 132 (Pt 11): 3175–3186. doi:10.1093/brain/awp236. ISSN 1460-2156. PMC 4038491. PMID 19767415.
- ^ Lampe, Anne Katrin; Flanigan, Kevin M.; Bushby, Katharine Mary; Hicks, Debbie (1993). "Collagen VI-Related Dystrophies". In Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.). Collagen Type VI-Related Disorders. Seattle (WA): University of Washington, Seattle. PMID 20301676. Retrieved 2020-10-19.
{{cite book}}:|work=ignored (help)