Tumor necrosis factor






Tumor necrosis factor (TNF), formerly known as TNF-α, is a chemical messenger produced by the immune system that induces inflammation.[5] TNF is produced primarily by activated macrophages, and induces inflammation by binding to its receptors on other cells.[6] It is a member of the tumor necrosis factor superfamily, a family of transmembrane proteins that are cytokines, chemical messengers of the immune system.[7] Excessive production of TNF plays a critical role in several inflammatory diseases, and TNF-blocking drugs are often employed to treat these diseases.[8]
TNF is produced primarily by macrophages but is also produced in several other cell types, such as T cells, B cells, dendritic cells, and mast cells. It is produced rapidly in response to pathogens, cytokines, and environmental stressors.[9] TNF is initially produced as a type II transmembrane protein (tmTNF), which is then cleaved by TNF alpha converting enzyme (TACE) into a soluble form (sTNF) and secreted from the cell.[10] Three TNF molecules assemble together to form an active homotrimer, whereas individual TNF molecules are inert.[10]
When TNF binds to its receptors, tumor necrosis factor receptor 1 (TNFR1) and tumor necrosis factor receptor 2 (TNFR2), a pathway of signals is triggered within the target cell, resulting in an inflammatory response. sTNF can only activate TNFR1, whereas tmTNF can activate both TNFR1 and TNFR2,[6] as well as trigger inflammatory signaling pathways within its own cell.[11] TNF's effects on the immune system include the activation of white blood cells, blood coagulation, secretion of cytokines, and fever.[5] TNF also contributes to homeostasis in the central nervous system.[12]
Inflammatory diseases such as rheumatoid arthritis, psoriasis, and inflammatory bowel disease can be effectively treated by drugs that inhibit TNF from binding to its receptors.[8] TNF is also implicated in the pathology of other diseases including cancer, liver fibrosis, and Alzheimer's, although TNF inhibition has yet to show definitive benefits.[13]
History
[edit]In the 1890s, William Coley observed that acute infections could cause tumor regression, leading to his usage of bacterial toxins as a cancer treatment. In 1944, endotoxin was isolated from Coley's bacterial toxins as the substance responsible for the anticancer effect. In particular, endotoxin could cause tumor regression when injected into mice with experimentally induced cancers. In 1975, Carswell et al. discovered that endotoxin did not directly cause tumor regression, but instead induced macrophages to secrete a substance that causes tumors to hemorrhage and necrotize, termed "tumor necrosis factor."[6]
In the 1980s, TNF was purified, sequenced, and cloned in bacteria. Studies on recombinant TNF confirmed the anticancer potential of TNF, but this optimism faded when TNF injections were found to induce endotoxin shock. TNF was also discovered to be the same protein as cachectin, known to cause muscle wasting in mice. These findings demonstrated that TNF could be detrimental in excessive quantities. In 1992, TNF antibodies were found to reduce joint inflammation in mice, revealing TNF's role in inflammatory diseases. This led to the approval of the first anti-TNF therapy for rheumatoid arthritis in 1998.[6]
Nomenclature
[edit]In 1985, TNF was found to have significant sequential and functional similarity with lymphotoxin, a previously discovered cytokine. This led to the renaming of TNF to TNF-α and lymphotoxin to TNF-β. However, in 1993, a protein with close similarity to lymphotoxin was discovered, termed lymphotoxin-β. In 1998, at the Seventh International TNF Congress, TNF-β was officially renamed to lymphotoxin-α, while TNF-α was renamed back to TNF. Nevertheless, some papers continue to use the term TNF-α.[14]
Evolution
[edit]The TNF and lymphotoxin-α genes are believed to be descended from a common ancestor gene that developed early in vertebrate evolution, before the Agnatha and Gnathostomata split. This ancestor gene was dropped from the Agnatha ancestor but persisted in the Gnathostomata ancestor. During the evolution of gnathostomes, this ancestor gene was duplicated into the TNF and lymphotoxin-α genes.[15] Thus, while the ancestor gene is found across a variety of gnathostome species, only a subset of gnathostome species contain a TNF gene. Some fish species, such as Danio, have been found to contain duplicates of the TNF gene.[15]
The TNF gene is very similar among mammals, ranging from 233 to 235 amino acids.[16] The TNF proximal promoter region is also highly conserved among mammals, and nearly identical among higher primates.[9] The similarity of the TNF gene among fish is lower, ranging from 226 to 256 amino acids. Like mammalian TNF, the fish TNF gene has been shown to be stimulated in macrophages by antigens.[16] All TNF genes have a highly conserved C-terminal module known as the TNF homology domain, due to its important role in binding TNF to its receptors.[15]
Gene
[edit]Location
[edit]The human TNF gene is mapped to chromosome 6p21.3, residing in the class III region of the major histocompatibility complex, where many immune system genes are contained. The class III region is sandwiched between the HLA-DR locus on the centromeric side, and the HLA-B locus on the telomeric side. The TNF gene is 250 kilobases away from the HLA-B locus, and 850 kilobases away from the HLA-DR locus. The TNF gene is located 1,100 kilobases downstream of the lymphotoxin-α gene.[17]
Expression
[edit]
TNF is produced rapidly in response to many stimuli by multiple cell types. Cell types that express TNF include T cells, B cells, macrophages, mast cells, dendritic cells, and fibroblasts, and stimuli that activate the TNF gene include pathogenic substances, cytokines from other immune cells, and environment stressors. A few such cytokines include interleukin-1, interleukin-2, interferon-γ, and TNF itself. TNF transcription is activated by a variety of signaling pathways and transcription factors, depending on the cell type and stimulus. TNF transcription does not depend on the synthesis of new proteins, enabling rapid activation of the gene.[9]
TNF gene expression is regulated by a proximal promoter region consisting of approximately 200 base pairs. Most of the binding sites within the proximal promoter region can recognize multiple transcription factors, enabling TNF to be activated by a variety of signaling pathways. As transcription factors bind to the promoter region, they also bind to coactivators, assembling into a large structure known as an enhanceosome. The composition of the enhanceosome depends on ambient factors within the cell, particularly nuclear factor of activated T-cells (NFAT).[9]
TNF expression is also regulated by DNA structure. DNA is coiled around histones, which is loosened by acetylation and condensed by methylation. Proteins that acetylate histones at the TNF promoter, particularly CREB-binding protein in T cells, are often critical for TNF expression. In contrast, several cell types that do not express TNF are highly methylated at the histones of the TNF promoter. Long-range intrachromosomal interactions can also regulate TNF expression. In activated T-cells, the DNA surrounding the TNF promoter circularizes, bringing promoter complexes closer together and enhancing transcription efficiency.[9]
Transcription
[edit]
The transcribed region contains 4 exons separated by 3 introns, for a total of 2,762 base pairs in the primary transcript and 1,669 base pairs in the mRNA.[19] The mRNA consists of four regions: the 5' untranslated region, which is not included in the TNF protein; the transmembrane portion, which is present in transmembrane TNF but not in soluble TNF; the soluble portion; and the 3' untranslated region. More than 80% of the soluble portion is contained in the last exon, while the transmembrane portion is contained in the first two exons. The 3' untranslated region contains an AU-rich element (ARE) that regulates the translation of TNF.[20] In unstimulated macrophages, various proteins bind to the ARE to destabilize TNF mRNA, suppressing the translation of TNF. Upon activation, TNF translation is unsuppressed.[21]
Protein
[edit]
TNF is initially produced as a transmembrane protein (tmTNF) consisting of 233 amino acids. tmTNF binds to both TNFR1 and TNFR2, but its activity is primarily mediated by TNFR2. Upon binding to a receptor, tmTNF also activates signaling pathways within its own cell. tmTNF is cleaved by TNF alpha converting enzyme (TACE), which causes the extracellular portion to be secreted. After cleavage, the remaining tmTNF is cleaved again by SPPL2B, causing the intracellular portion to translocate to the nucleus. There, it is believed to regulate cytokine production, such as triggering the expression of interleukin-12.[10]
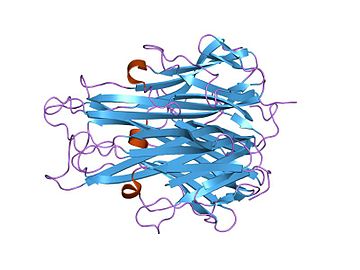
The secreted extracellular portion, denoted sTNF, consists of 157 amino acids.[22] Unlike tmTNF, sTNF can only bind to TNFR1.[10] The secondary structure of sTNF consists primarily of alternating strands that join into two sheets, known as antiparallel β-sheets. The two sheets are layered on top of each other, forming a wedge shape known as an antiparallel β-sandwich. Remarkably, this structure is similar to those seen on the coats of viruses. The last 9 residues of the C-terminus are locked into the middle strand of the bottom sheet, and are necessary for bioactivity.[22]
Both tmTNF and sTNF are only bioactive as homotrimers, whereas individual monomers are inactive.[10] The rate at which TNF trimers disassemble is constant, whereas the rate at which TNF trimers assemble increases with TNF concentration. This causes TNF to be mostly trimers at high concentrations, whereas TNF is mostly monomers and dimers at low concentrations.[23] The coexistence of TNF dimers and trimers in dynamic equilibrium suggests that TNF might be a morpheein.[24] Small molecules that stabilize TNF dimers and prevent the assembly of TNF trimers present a potential mechanism for inhibiting TNF.[25]
Function
[edit]TNF is a central mediator of the body's innate immune response.[5] By binding to receptors TNFR1 and TNFR2, TNF can induce either cell survival or cell death in a target cell. The cell survival response includes cell proliferation and the activation of inflammatory signals, while the cell death response can either be apoptosis, the controlled death of the cell, or necroptosis, a less controlled death causing inflammation and interference in surrounding tissue. TNF induces cell survival by default, but cell death can be induced by factors such as disruption of inflammatory pathways by pathogens, co-stimulation with other cytokines, and cross-talk between TNFR1 and TNFR2.[6] Additionally, transmembrane TNF (tmTNF) acts as a reverse signaler, triggering a variety of responses in its own cell depending on cell type and stimulant.[26]
TNFR1 signaling
[edit]
TNFR1 exists in most cell types and binds to both tmTNF and sTNF. TNFR1 contains a death domain in its cytoplasmic tail, enabling it to trigger cell death.[26] Whether TNFR1 activation triggers cell survival or cell death is mediated by the formation of protein complexes: complex I, which leads to cell survival, and complex II, which leads to cell death. By default, TNFR1 activation triggers cell proliferation and inflammation rather than cell death. These inflammatory pathways contain three cell death checkpoints, each of which is critical in preventing cell death.[6]
Upon activation by TNF, TNFR1 trimerizes and forms complex I by recruiting RIPK1 and TRADD, which recruits TRAF2, cIAP1 and cIAP2, and LUBAC. cIAP1 and cIAP2 are ubiquitin ligases that form K63-linked ubiquitin chains, which recruit TAK1 via TAB2 and TAB3. LUBAC is also a ubiquitin ligase that forms M1-linked ubiquitin chains, which attract IKK via NEMO. TAK1 activates the MAPK pathways, as well as IKK, which in turn activates the canonical NF-κB pathway. The MAPK pathways and the NF-κB pathway activate multiple transcription factors in the nucleus, which result in cell survival, proliferation, and inflammatory response. Complex I is negatively regulated by deubiquitinases such as A20, CYLD, and OTULIN, which destabilize complex I.[6]
Complex II is formed when RIPK1 and/or TRADD disassociate from complex I and bind with FADD to activate caspase 8, leading to cell death. Complex IIa includes TRADD and can activate caspase 8 without RIPK1, while complex IIb does not include TRADD, so it is dependent on RIPK1 for the activation of caspase 8. The pathways of complex I induce three checkpoints that prevent complex II from inducing cell death.[6]
In the first checkpoint, IKK disables RIPK1 via phosphorylation while it is attached to complex I. This disables complex IIb, which is dependent on RIPK1. Since IKK is dependent on the ubiquitination of complex I, conditions that affect ubiquitination, such as inhibition of cIAP1/2 and LUBAC, mutation of the RIPK1 ubiquitin acceptor site, or deficiencies of A20 and OUTLIN, can disable this checkpoint. The disabling of the IKK checkpoint activates complex IIb, leading to apoptosis, or pyroptosis by cleaving GSDMD. The disabling of the IKK checkpoint can also indirectly activate complex IIa by disabling the NF-κB pathway, which controls the second checkpoint.[6]
In the second checkpoint, the NF-κB pathway promotes the expression of pro-survival genes such as FLIP, which counteracts the activation of caspase 8 in complex IIa. This checkpoint can be disabled by translation inhibitors such as cycloheximide, as well as by the disabling of the IKK complex, which controls the NF-κB pathway. The disabling of this checkpoint activates complex IIa, leading to apoptosis.[6]
In the third checkpoint, non-lethal caspase 8 is activated by TNFR1 signalling, which binds to complex IIb and cleaves RIPK1, disabling it. It is unknown why this form of caspase 8 does not cause cell death. The disabling of this checkpoint, via inactivation of caspase 8, causes RIPK1 from complex IIb to bind to RIPK3 and MLKL, forming complex IIc, also referred to as the necrosome. The necrosome then causes necroptosis.[6]
TNFR2 signaling
[edit]
Unlike TNFR1, TNFR2 is expressed in limited cell types, including endothelial cells, fibroblasts, and subsets of neurons and immune cells. TNFR2 is only fully activated by tmTNF, while activation by sTNF is partially inhibited. Unlike TNFR1, TNFR2 does not possess a death domain, so it is incapable of directly inducing cell death. Thus, TNFR2 activation most often leads to cell survival. Cell survival can either lead to an inflammatory response, via canonical NF-κB activation, or cell proliferation, via non-canonical NF-κB activation, depending on intracellular conditions and the signaling process of TNFR1. TNFR2 can also indirectly cause cell death by disrupting the cell death checkpoints of TNFR1.[26]
Upon binding to tmTNF, TNFR2 trimerizes and directly recruits TRAF2, as well as TRAF1 or TRAF3. TRAF2 is central to the TNFR2 signaling complex and recruits cIAP1/2. If there is an accumulation of NIK within the cell, TRAF2/3 and cIAP1/2 may be formed as a complex with inactive NIK. When TRAF2/3 binds to TNFR2, the attached NIK is activated, which in turn activates IKKα. This allows p100 and RelB to be processed into a heterodimer which activates the non-canonical NF-κB pathway, leading to cell proliferation. The expression of p100 and RelB is potentiated by the activation of the canonical NF-κB pathway by TNFR1. Thus, TNFR2 non-canonical NF-κB activation is dependent on the canonical NF-κB activation by TNFR1, as well as the accumulation of NIK within the cell.[26]
TNFR2 can also activate the canonical NF-κB pathway, though this is less common than non-canonical NF-κB activation. The details of TNFR2's activation of the canonical NF-κB pathway are unknown. Presumably, TAK1 and IKK are recruited by the TRAF2 / TRAF1/3 / cIAP1/2 signalling complex, which in turn activates the canonical NF-κB pathway.[26]
TNFR2 can indirectly induce cell death by degrading cIAP1/2 as part of the non-canonical NF-κB pathway. The degradation of cIAP1/2 affects the ubiquitination of the TNFR1 signalling complex, which inhibits the function of IKK. This disables the IKK cell death checkpoint in TNFR1, inducing cell death.[6]
Reverse signalling
[edit]tmTNF can act as a receptor, activating pathways within its own cell upon binding to TNFR1 or TNFR2. tmTNF reverse signalling can induce apoptosis, apoptosis resistance, inflammation, or inflammation resistance depending on the ligand and cell type.[11]
In tumor cells, such as B lymphoma cells, tmTNF reverse signalling has been shown to increase NF-κB activity, enhancing cell survival and apoptosis resistance. In natural killer cells, tmTNF reverse signalling increases cytotoxic activity by increasing the expression of perforin, granzyme B, Fas ligand, and TNF. In T cells, the activation of the JNK pathway by tmTNF reverse signalling can lead to cell cycle inhibition and apoptosis.[11]
In monocytes, tmTNF has been shown to play a dual role in mediating the monocyte's inflammatory response to sTNF. If tmTNF reverse signalling occurs before a monocyte is activated by sTNF, then the monocyte's inflammatory response to sTNF is enhanced. If tmTNF reverse signalling occurs after a monocyte is activated by sTNF, then the inflammatory response is reduced.[11] Meanwhile, tmTNF reverse signalling reduces a monocyte's inflammatory response to endotoxin. This effect is caused by tmTNF activating the JNK and p38 pathways, which induces TGF-β production, which then interferes with the signalling pathway of endotoxin.[11]
Immune response
[edit]The innate immune system is the immune system's first line of defense, responding rapidly and nonspecifically to invading pathogens. It is activated when pathogen-associated molecular patterns (PAMPs), such as endotoxins and double-stranded viral RNA, bind to the pattern recognition receptors (PRRs) of immune cells, causing them to secrete immune-regulating cytokines. These cytokines, such as IL-1, IL-6, IL-8, and TNF, are primarily secreted by immune cells that engulf bacteria, such as macrophages and dendritic cells. They mainly act on white blood cells, as well as on endothelial cells in blood vessels to promote an early inflammatory response.[5]
TNF is the principal cytokine for regulating acute inflammation, though many of its functions are shared with other cytokines, especially IL-1. By binding to TNF receptors, TNF can perform functions including stimulating endothelial cells to induce coagulation, which obstructs blood flow to prevent the spread of microbes; stimulating endothelial cells and macrophages to secrete chemokines that attract white blood cells; stimulating the secretion of other cytokines such as IL-1; activating neutrophils and macrophages; stimulating the liver to produce acute phase proteins, such as C-reactive protein; inducing catabolism of muscles and fat to produce energy; and stimulating scar tissue formation, also known as fibrosis. In addition to inducing the secretion of cytokines, TNF itself can be induced by cytokines, enabling a cascade of inflammatory signals. Excessive amounts of TNF can cause septic shock.[5]
Much of TNF's functions are mediated through inflammatory signalling pathways, such as MAPK and NF-κB. Many pathogens attempt to prevent an immune response by hijacking cells and disrupting their inflammatory pathways. In response to this, the TNFR1 signalling pathway has cell death pathways that are inhibited by the activities of the inflammatory pathways. If a cell's inflammatory pathways are disrupted, the cell death pathways are uninhibited, triggering cell death. This prevents the pathogen from replicating within the cell, as well as alerting the immune system.[6]
Additionally, TNF induces fever to help the body fight infections. TNF can induce fever by triggering the release of cytokines interleukin-1 and interleukin-6, or through other mediators like PLA2. TNF or its mediators can reach the hypothalamus either through circulation in the bloodstream or through secretion by macrophages and endothelial cells near the hypothalamus. TNF can also induce fever by stimulating the primary vagal terminals in the liver, which signals to neurons to secrete norepinephrine. All of these pathways culminate in the synthesis of prostaglandins, which interact with the OVLT in the hypothalamus to raise the target temperature of the body.[27]
Central nervous system
[edit]TNF is expressed in various cells in the central nervous system, including glial cells, microglia, astrocytes, and neurons, and plays a critical role in maintaining homeostasis.[12]
Through TNFR1 signalling, TNF can increase the surface expression of AMPA receptors and NDMA receptors in neurons, strengthening synaptic transmission. TNF also decreases the surface expression of GABAA receptors, reducing the activity of inhibitory synapses. TNF can also modulate the release of glutamate, an excitatory neurotransmitter, and S100B, a zinc-binding protein, by astrocytes. The modulation of excitation and inhibition of neurons by TNF indicates that TNF plays a role in synaptic scaling and plasticity.[12]
Through TNFR2 signalling, TNF promotes the proliferation and maturation of oligodendrocytes, which produce protective myelin sheaths around nerve cells. On the other hand, TNF becomes cytotoxic to oligodendrocyte progenitor cells when the cells are in contact with astrocytes.[12]
Clinical significance
[edit]Autoimmunity
[edit]Excessive production of TNF plays a key role in the pathology of autoimmune diseases, such as rheumatoid arthritis, inflammatory bowel disease, psoriatic arthritis, psoriasis, and noninfectious uveitis.[8] In these diseases, TNF is erroneously secreted by immune cells in response to environmental factors or genetic mutations. TNF then triggers an inflammatory response, damaging normal tissue. TNF blockers, which prevent TNF from binding to its receptors, are often used to treat these diseases.[6]
TNF induces inflammation both by activating inflammatory pathways, as well as by triggering cell death. Cell death triggers inflammation by exposing the components of dying cells to neighboring cells, as well as by compromising barrier integrity in the skin and intestine, allowing microbes to infiltrate the tissue. TNF is believed to trigger cell death in inflammatory diseases due to elevated levels of interfering cytokines, elevated levels of TNFR2 signalling, or genetic mutations. Drugs that target proteins involved in TNF-induced cell death, such as RIPK1, are being evaluated for their efficacy against autoinflammatory diseases.[6]
Cancer
[edit]TNF was initially discovered as an agent that kills tumors, particularly sarcomas. However, TNF is now known to play a dual role in cancer, both as a promoter and inhibitor, due to its ability to induce either proliferation or death in tumor cells. The exact mechanisms determining which role TNF plays in cancer are unclear. In general, TNF is considered to be a cancer promoter.[28]
In some cancers, TNF has been shown to play an inhibitory role, primarily when injected locally, repeatedly, and at high concentrations. Due to TNF's adverse side effects, potential TNF cancer treatments seek to maximize cytotoxicity to tumors while minimizing exposure to the entire body. Some treatments increase cytotoxicity by inhibiting the cell survival pathways of tumors before treatment with TNF. Other treatments localize TNF activity using antibody-TNF fusions, also known as immunocytokines. Local TNF treatment has been shown to induce tumor regression, though they rarely induce complete remission. Body-wide administration of TNF has shown low efficacy and high side effects.[28]
In many cancers, TNF is believed to play a supportive role. High TNF expression levels are associated with more advanced cancers, and TNF expression is found in tumor cells from the early stages of disease. TNF expression can lead to the recruitment of white blood cells that promote metastasis, as well as direct activation of pathways that promote tumor survival, invasion, and metastasis. TNF-blockers such as infliximab and etanercept did not induce a response in most advanced or metastatic cancers, but some studies have shown disease stabilization.[28]
Infections
[edit]TNF plays a critical role in the innate immune response to infections. Accordingly, the use of TNF blockers is associated with increased risks of infection, such as with Varicella-zoster virus, Epstein–Barr virus, and Cytomegalovirus.[29]
Conversely, TNF plays a role in the progression of HIV by inducing apoptosis of T cells in HIV-infected people. TNF blockage has reportedly led to clinical improvement in HIV without worsening the infection, though data is limited.[29]
Sepsis
[edit]TNF is believed to be an important contributor to sepsis due to its ability to upregulate the innate immune system and blood coagulation. In animals, the injection of TNF can produce heart, lung, kidney, and liver dysfunction similar to sepsis. However, in humans suffering from sepsis, TNF is not consistently elevated.[30]
Although TNF blockers showed efficacy in treating sepsis in mice, they showed mixed results in treating sepsis in humans. This is believed to be due to the dual role that TNF plays in the immune system; blocking TNF reduces the severe inflammation that causes sepsis, but also hinders the immune system's ability to resist the infection. It is hypothesized that TNF blockers are more beneficial in cases of severe sepsis, where the probability of death is higher.[30]
Liver fibrosis
[edit]TNF is a key player in liver injury and inflammation, but its role in liver fibrosis is controversial. TNF contributes to the activation and survival of hepatic stellate cells (HSCs), believed to be the primary contributors of liver fibrosis. On the other hand, TNF suppresses alpha-1 type-1 collagen expression and HSC proliferation in vitro, which should inhibit liver fibrosis. In general, TNF is considered to promote liver fibrosis by promoting HSC survival.[31] Despite this connection, TNF blockers are not used to treat liver fibrosis. In clinical trials of alcoholic hepatitis, TNF blockers had no significant effect.[31]
Additionally, hepatocyte death, the initial event that drives liver injury and fibrosis, may be induced by TNF, though this connection is uncertain. TNF injection alone does not induce hepatocyte death in vivo. However, when TNF injection is coupled with survival pathway inhibition, such as during hepatitis C virus infection, TNF induces hepatocyte death and acute liver failure. The remnants of dead hepatocytes are consumed by HSCs and Kupffer cells, which then secrete fibrosis-promoting factors, such as TGF-β, as well as promoting further hepatocyte death.[31]
Insulin resistance
[edit]TNF promotes insulin resistance by inhibiting insulin receptor substrate 1 (IRS1). Under normal circumstances, IRS1, upon activation by insulin, undergoes tyrosine phosphorylation and increases glucose uptake in the cell. This process is disrupted when TNF induces the serine phosphorylation of IRS1, converting IRS1 into an insulin inhibitor. TNF-induced insulin resistance is common in cases of obesity and can lead to Type II Diabetes. TNF has been found to be upregulated in the adipose tissue of humans and animals with obesity, though it remains unclear why obesity induces high TNF levels.[32]
Nonalcoholic fatty liver disease
[edit]TNF plays a key role in nonalcoholic fatty liver disease (NAFLD), in which fat builds up in the liver, leading to injury, inflammation, and scarring. TNF promotes insulin resistance, which promotes fat build up in the liver. As fat builds up in the liver and surrounding adipose tissue, immune cells may infiltrate the expanding tissue and secrete TNF, causing inflammation. Thus, TNF may serve as a causal link between inflammation, insulin resistance, and fat accumulation in the liver. Clinical studies have shown that TNF levels are correlated with the severity of NAFLD, although some studies have shown otherwise. Pharmacological strategies that downregulate TNF have shown favorable effects on NAFLD, while the efficacy of TNF blockers is yet to be evaluated.[33]
Muscle wasting
[edit]Conditions that cause inflammation, such as cancer, can elevate TNF levels, which contributes to muscle wasting. TNF contributes to muscle wasting by activating the NF-κB pathway, which activates the ubiquitin–proteasome pathway to degrade protein, and by inhibiting the activation of satellite cells, which are responsible for protein regeneration. However, TNF blockers have had limited effect on muscle wasting in clinical studies, likely due to the multifactorial nature of muscle wasting.[34]
Exercise
[edit]During exercise, the level of IL-6, a TNF inhibitor, rapidly increases, leading to an anti-inflammatory effect. This is followed by a subsequent increase in the levels of IL-10 and soluble TNF receptors, both of which also inhibit TNF. While moderate exercise does not increase TNF levels, strenuous exercise has been shown to increase TNF levels two-fold, causing a pro-inflammatory effect. However, this proinflammatory effect is outweighed by the anti-inflammatory effect of IL-6, which can increase 50-fold. Regular exercise has been shown to reduce base TNF levels in the long term. Thus, exercise is generally considered to inhibit TNF, which contributes to the overall anti-inflammatory effect of exercise.[35]
Neuroinflammation
[edit]In the central nervous system, TNF is primarily produced by microglia, a type of macrophage, but also by neurons, endothelial cells, and immune cells. Excessive TNF contributes to neuroinflammation by causing excitotoxic neuronal cell death, increasing glutamate levels, activating microglial cells, and disrupting the blood–brain barrier. As a result, TNF is seen to play an important role in central nervous system disorders associated with neuroinflammation, including neurosarcoidosis, multiple sclerosis, Neuro-Behçet's disease.[36]
Paradoxically, TNF-blockers can cause demyelination of neurons and worsen multiple sclerosis symptoms. This is believed to be due to the homeostatic role of TNF in the central nervous system, especially on neuron myelination via TNFR2. The selective blockade of TNFR1 has shown positive outcomes in animal models.[36]
TNF-induced neuroinflammation has also been associated with Alzheimer's disease, and is suspected to contribute to the amyloid-β plaques and tau protein hyperphosphorylation found in the brains of Alzheimer's patients. TNF blockers have been associated with reduced risk of developing Alzheimer's. Some studies have shown TNF blockers to slightly improve cognition in Alzheimer's patients, though larger studies are needed. Since TNF blockers cannot pass through the blood–brain barrier, it is believed that reducing TNF levels across the body also reduces TNF levels within the brain.[37]
TRAPS
[edit]In TNF receptor associated periodic syndrome (TRAPS), genetic mutations in TNFR1 lead to defective binding of TNFR1 to TNF, as well as defective shedding of TNFR1, a mechanism that attenuates TNFR1 signalling. This causes periodic inflammation, though the exact mechanism is unknown. TNF blockers such as etanercept have shown partial efficacy in reducing symptoms, while other TNF blockers such as adalimumab and infliximab have been shown to worsen symptoms.[38]
Taste perception
[edit]Excessive levels of inflammatory cytokines, such as during infection or autoimmunity, have been associated with anorexia and reduced food intake. It is hypothesized that TNF reduces food intake by increasing sensitivity to bitter taste, though the exact mechanisms of this are unknown.[39]
Pharmacology
[edit]TNF blockers
[edit]TNF blockers bind to TNF to prevent it from activating its receptors. Additionally, TNF blockers that bind to tmTNF may induce apoptosis in TNF-expressing cells, eliminating inflammatory immune cells.[36] TNF blockers can be monoclonal antibodies, such as infliximab, while others are decoy fusion proteins, like etanercept.[8] New TNF blockers are being developed, including small compounds that can specifically target TNF and monoclonal antibodies with lower immunogenicity potential.[8] Rarely, the suppression of TNF can lead to the development of a new form of "paradoxical" autoimmunity, caused by the overexpression of other cytokines.[40]
References
[edit]- ^ a b c ENSG00000230108, ENSG00000223952, ENSG00000204490, ENSG00000228321, ENSG00000232810, ENSG00000228849, ENSG00000206439 GRCh38: Ensembl release 89: ENSG00000228978, ENSG00000230108, ENSG00000223952, ENSG00000204490, ENSG00000228321, ENSG00000232810, ENSG00000228849, ENSG00000206439 – Ensembl, May 2017
- ^ a b c GRCm38: Ensembl release 89: ENSMUSG00000024401 – Ensembl, May 2017
- ^ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ a b c d e Kaiser G (21 November 2013). "11.3C: Cytokines Important in Innate Immunity". Microbiology. LibreTexts.
- ^ a b c d e f g h i j k l m n o p van Loo G, Bertrand MJ (May 2023). "Death by TNF: a road to inflammation". Nature Reviews Immunology. 23 (5): 289–303. doi:10.1038/s41577-022-00792-3. PMC 9665039. PMID 36380021.
- ^ Croft M, Siegel RM (March 2017). "Beyond TNF: TNF superfamily cytokines as targets for the treatment of rheumatic diseases". Nature Reviews Rheumatology. 13 (4): 217–233. doi:10.1038/nrrheum.2017.22. PMC 5486401. PMID 28275260.
- ^ a b c d e Jang DI, Lee AH, Shin HY, Song HR, Park JH, Kang TB, et al. (March 2021). "The Role of Tumor Necrosis Factor Alpha (TNF-α) in Autoimmune Disease and Current TNF-α Inhibitors in Therapeutics". International Journal of Molecular Sciences. 22 (5): 2719. doi:10.3390/ijms22052719. PMC 7962638. PMID 33800290.
- ^ a b c d e f Falvo JV, Tsytsykova AV, Goldfeld AE (2010). "Transcriptional control of the TNF gene". TNF Pathophysiology. Current Directions in Autoimmunity. Vol. 11. Karger. pp. 27–60. doi:10.1159/000289196. ISBN 978-3-8055-9384-7. PMC 4785889. PMID 20173386.
- ^ a b c d e Horiuchi T, Mitoma H, Harashima S, Tsukamoto H, Shimoda T (July 2010). "Transmembrane TNF-alpha: structure, function and interaction with anti-TNF agents". Rheumatology. 49 (7): 1215–1228. doi:10.1093/rheumatology/keq031. PMC 2886310. PMID 20194223.
- ^ a b c d e Szondy S, Pallai A (January 2017). "Transmembrane TNF-alpha reverse signaling leading to TGF-beta production is selectively activated by TNF targeting molecules: Therapeutic implications". Pharmacological Research. 115: 124–132. doi:10.1016/j.phrs.2016.11.025. PMID 27888159.
- ^ a b c d Caldito NG (July 2023). "Role of tumor necrosis factor-alpha in the central nervous system: a focus on autoimmune disorders". Frontiers in Immunology. 14. doi:10.3389/fimmu.2023.1213448. PMC 10360935. PMID 37483590.
- ^ Sethi JK, Hotamisligil GS (October 2021). "Metabolic Messengers: tumour necrosis factor". Nature Metabolism. 3 (10): 1302–1312. doi:10.1038/s42255-021-00470-z. PMID 34650277.
- ^ Grimstad Ø (May 2016). "Tumor Necrosis Factor and the Tenacious α". JAMA Dermatology. 152 (6): 557. doi:10.1001/jamadermatol.2015.4322. hdl:10037/10660. PMID 27168212.
- ^ a b c Marín I (November 2020). "Tumor Necrosis Factor Superfamily: Ancestral Functions and Remodeling in Early Vertebrate Evolution". Genome Biology and Evolution. 12 (11): 2074–2092. doi:10.1093/gbe/evaa140. PMC 7674686. PMID 33210144.
- ^ a b Goetz FW, Planas JV, MacKenzie SI (May 2004). "Tumor necrosis factors". Development & Comparative Immunology. 28 (5): 487–497. doi:10.1016/j.dci.2003.09.008. PMID 15062645.
- ^ Papadakis KA, Targan SR (October 2000). "Tumor necrosis factor: biology and therapeutic inhibitors". Gastroenterology. 119 (4): 1148–1157. doi:10.1053/gast.2000.18160. PMID 11040201.
- ^ Nedwin GE, Naylor SL, Sakaguchi AY, Smith D, Jarrett-Nedwin J, Pennica D, et al. (September 1985). "Human lymphotoxin and tumor necrosis factor genes: structure, homology and chromosomal localization". Nucleic Acids Research. 13 (17): 6361–6373. doi:10.1093/nar/13.17.6361. PMC 321958. PMID 2995927.
- ^ Chen F (June 2004). "TNF (tumor necrosis factor (TNF superfamily, member 2))". Atlas of Genetics and Cytogenetics in Oncology and Haematology.
- ^ Parameswaran N, Patial S (2010). "Tumor Necrosis Factor-α Signaling in Macrophages". Critical Reviews in Eukaryotic Gene Expression. 20 (2): 87–103. doi:10.1615/critreveukargeneexpr.v20.i2.10. PMC 3066460. PMID 21133840.
- ^ Zhang T, Kruys V, Huez G, Gueydan C (November 2002). "AU-rich element-mediated translational control: complexity and multiple activities of trans-activating factors". Biochemical Society Transactions. 30 (6): 952–958. doi:10.1042/bst0300952. PMID 12440953.
- ^ a b c Jones E, Stuart D, Walker N (March 1989). "Structure of tumour necrosis factor". Nature. 338 (6212): 225–228. Bibcode:1989Natur.338..225J. doi:10.1038/338225a0. PMID 2922050.
- ^ van Schie KA, Ooijevaar-de Heer P, Dijk L, Kruithof S, Wolbink G, Rispens T (September 2016). "Therapeutic TNF Inhibitors can Differentially Stabilize Trimeric TNF by Inhibiting Monomer Exchange". Scientific Reports. 6: 32747. Bibcode:2016NatSR...632747V. doi:10.1038/srep32747. PMC 5015024. PMID 27605058.
- ^ Lawrence SH, Jaffe EK (January 2013). "Expanding the Concepts in Protein Structure-Function Relationships and Enzyme Kinetics: Teaching using Morpheeins". Biochemistry and Molecular Biology Education. 36 (4): 274–283. doi:10.1002/bmb.20211. PMC 2575429. PMID 19578473.
- ^ Jaffe EK (January 2013). "Impact of quaternary structure dynamics on allosteric drug discovery". Current Topics in Medicinal Chemistry. 13 (1): 55–63. doi:10.2174/1568026611313010006. PMC 3631351. PMID 23409765.
- ^ a b c d e f Gough P, Myles IA (November 2020). "Tumor Necrosis Factor Receptors: Pleiotropic Signaling Complexes and Their Differential Effects". Frontiers in Immunology. 11. doi:10.3389/fimmu.2020.585880. PMC 7723893. PMID 33324405.
- ^ Netea MG, Kullberg BJ, Van der Meer JW (October 2000). "Circulating Cytokines as Mediators of Fever". Clinical Infectious Diseases. 31: 178–184. doi:10.1086/317513. PMID 11113021.
- ^ a b c Ben-Baruch A (May 2022). "Tumor Necrosis Factor α: Taking a Personalized Road in Cancer Therapy". Frontiers in Immunology. 13. doi:10.3389/fimmu.2022.903679. PMC 9157545. PMID 35663982.
- ^ a b Kim SY, Solomon DH (February 2010). "Tumor necrosis factor blockade and the risk of viral infection". Nature Reviews Rheumatology. 6 (3): 165–174. doi:10.1038/nrrheum.2009.279. PMC 3155180. PMID 20142812.
- ^ a b Qiu P, Cui X, Barochia A, Li Y, Natanson C, Eichacker PQ (November 2011). "The evolving experience with therapeutic TNF inhibition in sepsis: considering the potential influence of risk of death". Expert Opinion on Investigational Drugs. 20 (11): 1555–1564. doi:10.1517/13543784.2011.623125. PMC 3523300. PMID 21961576.
- ^ a b c Yang YM, Seki E (December 2015). "TNFα in liver fibrosis". Current Pathobiology Reports. 3 (4): 253–261. doi:10.1007/s40139-015-0093-z. PMC 4693602. PMID 26726307.
- ^ Bordon Y (June 2021). "TNF short-circuits the insulin receptor". Nature Milestones.
- ^ Vachliotis ID, Polyzos SA (July 2023). "The Role of Tumor Necrosis Factor-Alpha in the Pathogenesis and Treatment of Nonalcoholic Fatty Liver Disease". Current Obesity Reports. 12 (3): 191–206. doi:10.1007/s13679-023-00519-y. PMC 10482776. PMID 37407724.
- ^ Zhou J, Liu B, Liang C, Li Y, Song YH (May 2016). "Cytokine Signaling in Skeletal Muscle Wasting". Trends in Endocrinology & Metabolism. 27 (5): 335–347. doi:10.1016/j.tem.2016.03.002. PMID 27025788.
- ^ Docherty S, Harley R, McAuley JJ, et al. (January 2022). "The effect of exercise on cytokines: implications for musculoskeletal health: a narrative review". BMC Sports Science, Medicine and Rehabilitation. 14 (1): 5. doi:10.1186/s13102-022-00397-2. PMC 8740100. PMID 34991697.
- ^ a b c Caldito NG (July 2023). "Role of tumor necrosis factor-alpha in the central nervous system: a focus on autoimmune disorders". Frontiers in Immunology. 14. doi:10.3389/fimmu.2023.1213448. PMC 10360935. PMID 37483590.
- ^ Torres-Acosta N, O'Keefe JH, O'Keefe EL, Isaacson R, Small G (November 2020). "Therapeutic Potential of TNF-α Inhibition for Alzheimer's Disease Prevention". Journal of Alzheimer's Disease. 78 (2): 619–626. doi:10.3233/JAD-200711. PMC 7739965. PMID 33016914.
- ^ Cudrici C, Deuitch N, Aksentijevich I (May 2020). "Revisiting TNF Receptor-Associated Periodic Syndrome (TRAPS): Current Perspectives". International Journal of Molecular Sciences. 21 (9): 3263. doi:10.3390/ijms21093263. PMC 7246474. PMID 32380704.
- ^ Rogers N (April 2015). "Why illness might leave a bitter taste in the mouth". Nature. doi:10.1038/nature.2015.17415.
- ^ Lopetuso LR, Cuomo C, Mignini I, Gasbarrini A, Papa A (May 2023). "Focus on Anti-Tumour Necrosis Factor (TNF)-α-Related Autoimmune Diseases". International Journal of Molecular Sciences. 24 (9): 8187. doi:10.3390/ijms24098187. PMC 10179362. PMID 37175894.
External links
[edit]- Tumor Necrosis Factor-alpha at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- Overview of all the structural information available in the PDB for UniProt: P01375 (Tumor necrosis factor) at the PDBe-KB.
PDB gallery | |
|---|---|
|