Mulibrey nanism
| Mulibrey nanism | |
|---|---|
| Other names | Perheentupa syndrome and Pericardial constriction with growth failure[1] |
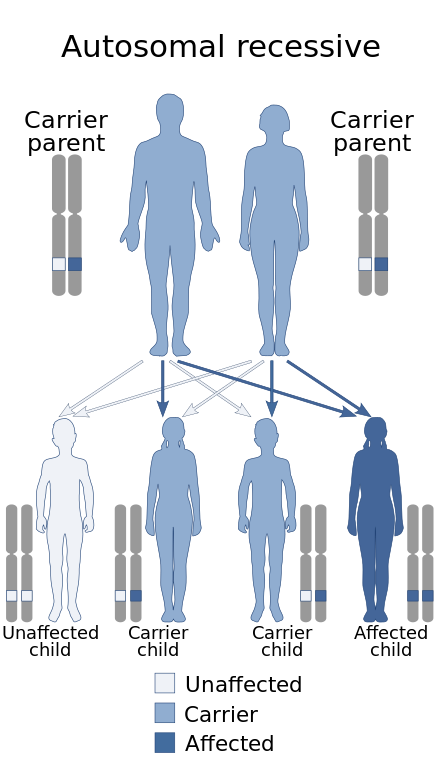 | |
| Mulibrey nanism has an autosomal recessive pattern of inheritance. | |
| Specialty | Rheumatology, medical genetics |
| Symptoms | Infertility[1] |
| Causes | Mutation of the TRIM37 gene[2] |
| Diagnostic method | Genetic testing[3] |
| Treatment | Growth hormone treatment, Regular pelvic exams[4] |
Mulibrey nanism ("MUscle-LIver-BRain-EYe nanism") is a rare autosomal recessive congenital disorder. It causes severe growth failure along with abnormalities of the heart, muscle, liver, brain and eye. TRIM37 is responsible for various cellular functions including developmental patterning.[5][6][7]
Signs and symptoms
[edit]An individual with Mulibrey nanism has growth retardation, a short broad neck, misshapen sternum, small thorax, square shoulders, enlarged liver, and yellowish dots in the ocular fundi.[1][7][8] Individuals with Mulibrey nanism have also been reported to have intellectual disability, tumors, and infertility.[1]
Genetics
[edit]
Mulibrey nanism is caused by mutations of the TRIM37 gene,[2] located at human chromosome 17q22-23.[9] The disorder is inherited in an autosomal recessive manner.[7] This means the defective gene responsible for the disorder is located on an autosome (chromosome 17 is an autosome), and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any symptoms of the disorder.[medical citation needed]
Diagnosis
[edit]The diagnosis of Mulibrey nanism can be done via genetic testing,[3] as well as by the physical characteristics (signs/symptoms) displayed by the individual.[10]
Treatment
[edit]
In terms of treatment/management for those with Mulibrey nanism should have routine medical follow-ups, additionally the following can be done:[4]
- Growth hormone treatment
- Regular pelvic exams
- Pericardiectomy
Prevalence
[edit]Worldwide, it has been documented in 110 persons, 85 of them Finnish. It is a recessive genetic disease. Many people with Mulibrey nanism have parents who are closely related, consanguine. Signs and symptoms are variable: siblings who suffer this disease sometimes do not share the same symptoms.[1]
See also
[edit]References
[edit]- ^ a b c d e OMIM (1966-2009). Mulibrey nanism. NCBI (Johns Hopkins University). Retrieved May 7, 2009 from, https://www.ncbi.nlm.nih.gov/omim/?term=253250
- ^ a b Reference, Genetics Home. "TRIM37 gene". Genetics Home Reference. Retrieved 2017-01-09.
- ^ a b "Mulibrey nanism - Tests - GTR - NCBI". www.ncbi.nlm.nih.gov. Retrieved 9 January 2017.
- ^ a b MD, Robert P. Erickson; PhD, Anthony J. Wynshaw-Boris MD (2016). Epstein's Inborn Errors of Development: The Molecular Basis of Clinical Disorders of Morphogenesis. Oxford University Press. p. 1415. ISBN 9780190275426. Retrieved 9 January 2017.
- ^ "TRIM37 tripartite motif-containing 37". NCBI Entrez Gene. Retrieved 2009-05-07.
- ^ RESERVED, INSERM US14 -- ALL RIGHTS. "Orphanet: MULIBREY nanism". www.orpha.net. Retrieved 2017-01-08.
{{cite web}}: CS1 maint: numeric names: authors list (link) - ^ a b c "Mulibrey Nanism | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 2017-01-08. Retrieved 2017-01-07.
- ^ Karlberg, N.; Jalanko, H.; Perheentupa, J.; Lipsanen-Nyman, M. (2004-02-01). "Mulibrey nanism: clinical features and diagnostic criteria". Journal of Medical Genetics. 41 (2): 92–98. doi:10.1136/jmg.2003.014118. ISSN 1468-6244. PMC 1735664. PMID 14757854.
- ^ Online Mendelian Inheritance in Man (OMIM): 605073
- ^ Disorders, the National Organization for Rare, ed. (2003). NORD guide to rare disorders. Philadelphia: Lippincott Williams & Wilkins. p. 94. ISBN 9780781730631. Retrieved 9 January 2017.
Further reading
[edit]- Traboulsi, Elias I., ed. (2011). Genetic Diseases of the Eye (2nd ed.). Oxford: Oxford University Press, USA. ISBN 9780199716975. Retrieved 9 January 2017.
- al.], Christopher J.H. Kelnar ... [et; Savage, Martin; Saenger, Paul; Cowell, Chris (2007). Growth disorders (2nd ed.). London: Hodder Arnold. ISBN 9780340812402.