半乳糖唾液酸贮积症
| 半乳糖唾液酸贮积症 | |
|---|---|
| 又称 | 神经氨酸酶缺乏症伴β-半乳糖苷酶缺乏症[1] |
 | |
| 溶酶体含有分解细胞废物的消化酶。 | |
| 分类和外部资源 | |
| 醫學專科 | 内分泌学 |
| ICD-10 | E77.1 |
| ICD-9-CM | 277.6 |
| OMIM | 256540、256540 |
| DiseasesDB | 33441 |
| Orphanet | 351 |
半乳糖唾液酸贮积症(英文:Galactosialidosis),也称为神经氨酸酶缺乏症伴β-半乳糖苷酶缺乏症,是一种遗传性溶小体储积症。[2]它是由CTSA基因突变而引起的,该突变导致β-半乳糖苷酶和神经氨酸酶的缺乏。这种缺乏会抑制细胞溶酶体的正常功能,导致细胞内有毒物质的积聚。标志性症状包括脊柱结构异常、视力问题、粗糙的面部特征、听力障碍和智力障碍。由于半乳糖唾液酸贮积症涉及所有细胞的溶酶体,所以它会影响身体的各个部位,包括大脑、眼睛、骨骼和肌肉。[3]根据患者出现症状时的年龄,该疾病由三种亚型组成:早期婴儿型、晚期婴儿型和青少年或成人型。[4]这种情况被认为是罕见的,大多数病例都发生在青少年或成人患者群体中。
体征和症状
[编辑]半乳糖唾液酸贮积症的体征和症状从轻微到严重不等,并且根据症状开始时个体的年龄以及患者所患疾病的特定亚型而有所不同。[4]患同一亚型的个体之间的症状也可能不同,但目前所知的是,一个人越早出现症状,随着时间的推移,疾病往往会发展得越严重。[來源請求]
被诊断患有早期婴儿型半乳糖唾液酸贮积症的个体会在出生前或出生后出现症状,而且往往最严重。最常见的症状包括体液异常增多、肝脾肿大、心脏增大、骨骼发育异常、肾脏疾病逐渐恶化、面部特征粗糙以及眼球后部出现樱桃红斑点。[5]
被诊断患有晚期婴儿半型乳糖唾液酸贮积症的个体的症状会与早期婴儿半乳糖唾液酸贮积症相似,但只是出现在6个月左右,而且往往较轻微。此亚型的其他症状可能包括生长问题、听力或视力问题以及癫痫发作。[5]
被诊断患有青少年或成人型半乳糖唾液酸贮积症的个体的症状包括平衡或协调障碍、皮肤上的暗红色斑点或血管角化瘤、视力丧失、癫痫发作和肌肉抽搐或肌阵挛。此亚型的症状往往不如前两种亚型严重,通常开始于青春期,患者平均年龄为16岁。[6]


成因
[编辑]分子生物学
[编辑]半乳糖唾液酸贮积症是由突变的CTSA基因引起的。该基因编码组织蛋白酶A,[7]它与神经氨酸酶1和β-半乳糖苷酶形成蛋白质复合体,分解溶酶体摄入的脂肪、糖和蛋白质。[4]在这个过程中需要组织蛋白酶A,因为在复合体中,它可以防止神经氨酸酶1和β-半乳糖苷酶过早分解,从而使它们可以正常发挥功能。当CTSA基因发生突变时,组织蛋白酶A的蛋白质结构发生缺陷,使其无法与其他蛋白质结合或形成复合体。这会导致β-半乳糖苷酶和神经氨酸酶1的缺乏。[8]因此,溶酶体无法分解有毒物质,从而让废物在细胞内堆积。[來源請求]

基因遗传
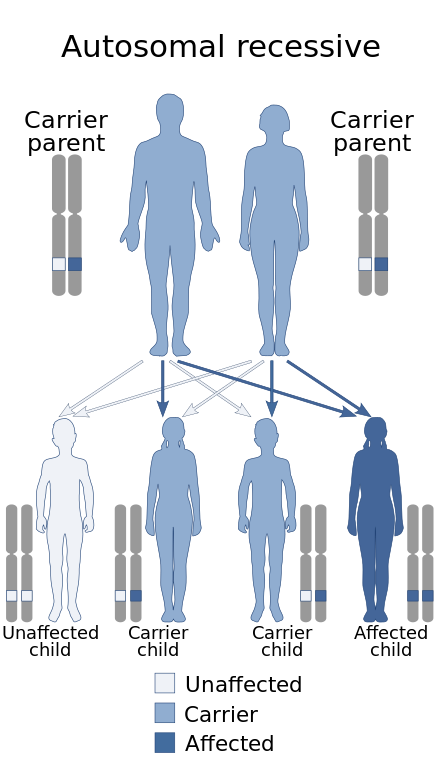
[编辑]所有被诊断患有半乳糖唾液酸贮积症的个体都以常染色体隐性方式遗传了该病症,无论年龄或亚型如何。所有基因均由两份拷贝组成,父母各一份。当一种疾病是隐性的时,这意味着患者需要一个基因的两个突变拷贝才能继承该特征或疾病。[9]当父母都携带同一个基因的一个突变拷贝时,他们的后代有25%的可能性会继承该基因的两个拷贝并患上这种疾病。在患有半乳糖唾液酸贮积症的人群中,患者的父母都携带CTSA基因的突变拷贝。受影响的孩子继承了这两个突变的基因,导致疾病的发展。[來源請求]
没有已知的环境或医学与半乳糖唾液酸贮积症的病因有关。
发病机制
[编辑]当患者遗传了两个突变的CTSA基因拷贝时,就会发生半乳糖唾液酸贮积症。突变基因的编码导致组织蛋白酶A的缺陷形式。当组织蛋白酶A的结构因突变而被破坏时,它将失去功能,不能与神经氨酸酶1和β-半乳糖苷酶形成消化复合物。最终,由于溶酶体无法发挥其功能,有毒物质会在人体细胞内积聚。[4]
诊断
[编辑]当怀疑是半乳糖唾液酸贮积症的特征性症状时,患者可以接受特定检测以确认诊断。其中一种常见的方法是测量神经氨酸酶1和β-半乳糖苷酶活性的酶活性分析。[4]酶活性水平降低表明缺乏组织蛋白酶A。可以进行完整的尿液分析以检测寡糖的存在。[4]由于溶酶体功能障碍,当寡糖在细胞内积累过量时,它会通过尿液排除体外。[來源請求]
此外,可以通过分子遗传分析更具体地确认诊断,分子遗传分析用于识别CTSA基因中的突变。一旦检测到突变,将检测结果与体检结果和患者的症状相结合,以全面诊断半乳糖唾液酸贮积症。[3]患者出现症状时的年龄用于确定患者的具体疾病亚型:[3]早期婴儿型患者从出生至3个月大之间被诊断出来、晚期婴儿型患者在3至12个月大之间被诊断出来、而青少年或成人型患者通常在症状开始出现的青春期被诊断出来。[10]
Kleijer等人于1979年通过测量培养的羊水细胞中的β-半乳糖苷酶和神经氨酸酶活性进行了产前诊断。[11]
预防
[编辑]由于这种疾病是遗传性的,因此唯一可用于预防这种疾病的方法是通过基因携带者筛查,即在父母决定要孩子之前检测其是否存在隐性突变的CTSA基因。如果发现父母双方都是突变基因的携带者,则孩子有25%的可能性遗传该病。遗憾的是这是无法避免的。[來源請求]
治疗
[编辑]目前,还没有治愈半乳糖唾液酸贮积症的方法。但是,有一些治疗方法可以帮助控制症状并提供支持性治療。[10]例如,治疗计划可能包括控制癫痫发作或肌肉抽搐的药物。患者根据他们正在经历的症状和出现的频率,与医学遗传学家、神经学家和眼科[3]医生建立常规护理也很常见。
预后
[编辑]被诊断患有半乳糖唾液酸贮积症的个体的预后因患者而异,这取决于年龄和症状的严重程度。因此,没有确定患者死亡或死亡风险增加的平均年龄。任何形式的疾病都没有完全康复的机会。[來源請求]
早期婴儿型
[编辑]被诊断为早期婴儿型的患者具有更严重、更具威胁性的症状,并且存活机会降低,因为这种亚型发展得更快,[4]但值得注意的是,大多数患者确实能活到婴儿晚期。[10]
晚期婴儿型
[编辑]由于晚期婴儿型患者在一岁内出现症状,病情没有早期婴儿亚型严重,生存机会更高,但患者的生活质量仍会受到很大影响,因为患者在出现症状时还很年轻。患者仍需要详细的护理计划来控制症状。[12]
青少年或成人型
[编辑]被诊断为青少年或成人型的患者症状较轻微,但必须处理渐进性精神障碍、脊柱畸形和癫痫等其他常见症状。然而,患有这种亚型的患者寿命通常与正常预期寿命相关。[13]
流行病学
[编辑]半乳糖唾液酸贮积症被认为是一种非常罕见的疾病,但其患病率尚不清楚。文献中记录的病例不到150例。在该人群中,约60%的病例属于青少年或成人型,[6]症状始于16岁或16岁之后。 据记载,大多数病例发生在拥有日本血统的人群中。[3]
研究
[编辑]当前需要进行研究,以便未来更好地了解半乳糖唾液酸贮积症、诊断进展和更有效的治疗。更多的研究已经完成,以确定半乳糖唾液酸贮积症患者的人口统计学和临床特征,最终目标是将这些信息用于基因转移治疗研究。[14]
目前至少有两项针对半乳糖唾液酸贮积症的临床试验正在进行。一项研究涉及对构成一些最罕见的溶酶体疾病的糖蛋白血症的研究,包括半乳糖唾液酸贮积症、天冬氨酰葡萄糖胺尿症、岩藻糖苷贮积症、辛德勒病和唾液酸沉积症等疾病。这是一项针对100名被诊断患有九种糖蛋白沉积症中的任何一种的患者的纵向研究。该研究的目的是更好地定义疾病的常见程度,确定可能有助于早期诊断的临床特征,详细说明疾病的进展,评估目前使用的支持疗法,并确定可能的治疗方法。[15]
参考文献
[编辑]- ^ RESERVED, INSERM US14-- ALL RIGHTS. Orphanet: Galactosialidosis. www.orpha.net. [11 April 2019]. (原始内容存档于2022-12-14) (英语).
- ^ Koike K, Hamaguchi T, Kitamura H, Imasawa T, Joh K. Galactosialidosis associated with IgA nephropathy: morphological study of renal biopsy. Pathol. Int. 2008, 58 (5): 295–9. PMID 18429828. S2CID 205477388. doi:10.1111/j.1440-1827.2008.02226.x.
- ^ 3.0 3.1 3.2 3.3 3.4 Galactosialidosis | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. rarediseases.info.nih.gov. [2020-10-14]. (原始内容存档于2022-12-11).
- ^ 4.0 4.1 4.2 4.3 4.4 4.5 4.6 Galactosialidosis. Rare Genomics Institute. [2020-10-14]. (原始内容存档于2022-12-11) (美国英语).
- ^ 5.0 5.1 Galactosialidosis | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. rarediseases.info.nih.gov. [2020-10-14]. (原始内容存档于2022-12-11).
- ^ 6.0 6.1 Galactosialidosis: MedlinePlus Genetics. medlineplus.gov. [2020-11-11]. (原始内容存档于2022-12-12) (英语).
- ^ Kleijer WJ, Geilen GC, Janse HC, et al. Cathepsin A deficiency in galactosialidosis: studies of patients and carriers in 16 families. Pediatr. Res. 1996, 39 (6): 1067–71. PMID 8725271. doi:10.1203/00006450-199606000-00022
.
- ^ Caciotti A, Catarzi S, Tonin R, Lugli L, Perez CR, Michelakakis H, Mavridou I, Donati MA, Guerrini R, D'Azzo A, Morrone A. Galactosialidosis: review and analysis of CTSA gene mutations. Orphanet J Rare Dis. 2013, 8 (1): 114. PMC 3737020
- ^ Autosomal Recessive Inheritance. www.stlouischildrens.org. [2020-11-11]. (原始内容存档于2022-12-12) (英语).
- ^ 10.0 10.1 10.2 Galactosialidosis. ISMRD. [2020-11-11]. (原始内容存档于2022-12-12) (英语).
- ^ Kleijer, W. J.; Hoogeveen, A.; Verheijen, F. W.; Niermeijer, M. F.; Galjaard, H.; O'Brien, J. S.; Warner, T. G. Prenatal diagnosis of sialidosis with combined neuraminidase and beta-galactosidase deficiency. Clinical Genetics. July 1979, 16 (1): 60–61. ISSN 0009-9163. PMID 477017. S2CID 41124809. doi:10.1111/j.1399-0004.1979.tb00851.x.
- ^ Galactosialidosis | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. rarediseases.info.nih.gov. [2020-10-14]. (原始内容存档于2022-12-11).
- ^ Galactosialidosis. ISMRD. [2020-11-11]. (原始内容存档于2022-12-12) (英语).
- ^ Characterization of the Patient Population With Galactosialidosis - Full Text View - ClinicalTrials.gov. clinicaltrials.gov. 9 October 2018 [2020-11-11]. (原始内容存档于2022-12-12) (英语).
- ^ Greenwood Genetic Center. Longitudinal Studies of the Glycoproteinoses. 2017-07-31 [2022-12-12]. (原始内容存档于2022-12-12).