Maladie de Charcot-Marie-Tooth
Pour les articles homonymes, voir CMT (homonymie).
Ne pas confondre avec la maladie de Charcot.
Les maladies de Charcot-Marie-Tooth, ou CMT, sont un ensemble de maladies neurologiques génétiques parmi les plus fréquentes. Ces maladies génétiques rares concernent environ 1 naissance sur 2 500 en France.
| Causes | Mutation |
|---|---|
| Symptômes | Faiblesse musculaire (en), fonte musculaire, pied creux, déficit sensoriel (en), hyporéflexie (en) et scoliose |
| Traitement | Inconnu (d) et physiothérapie |
|---|---|
| Spécialité | Neurologie |
| OMIM | 311860 |
|---|---|
| DiseasesDB | et 2343.htm 5815 et 2343 |
| MedlinePlus | 000727 |
| eMedicine | 1232386 |
| MeSH | D002607 |
Les CMT ne doivent pas être confondues avec la maladie de Charcot ou sclérose latérale amyotrophique qui est beaucoup plus grave.
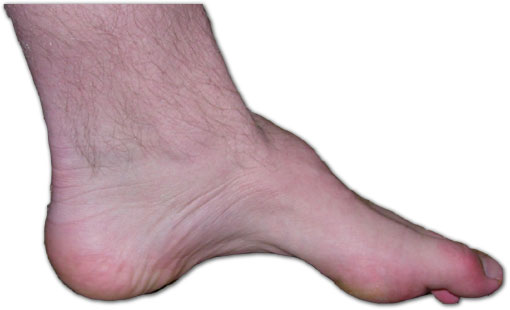
Décrite en 1886 par le neurologue français Jean-Martin Charcot et son étudiant Pierre Marie, puis par le neurologue britannique Howard Henry Tooth (en), la CMT est une neuropathie héréditaire sensitivo-motrice qui n’affecte pas l’espérance de vie et n’entraîne pas de retard mental. Elle touche indifféremment l’homme ou la femme. Schématiquement, la CMT est liée à l’atteinte des nerfs périphériques, reliant la moelle épinière aux muscles, ce qui perturbe la conduction de l’influx nerveux. Elle entraîne des troubles de la marche et une déformation fréquente des pieds (pieds creux). Cette maladie peut se déclarer dès l'enfance, mais également se développer assez tard, à l’âge adulte[1]. En général, la CMT évolue lentement mais elle peut aussi progresser par poussées.
Principaux symptômes
modifierLes principaux symptômes sont :
Traitements connus
modifierIl est d'abord fortement recommandé aux personnes atteintes de la maladie de Charcot Marie-Tooth d'être suivies par un médecin MPR - médecine physique et de réadaptation - et un neurologue. Le neurologue permettra de tenir le patient informé d'un potentiel traitement en plus de suivre l'évolution de la maladie. Le médecin MPR pourra prescrire des éléments apportant du confort pour le quotidien de ces personnes. Il s'agit [4]:
- des orthèses plantaires, des orthèses de nuit, un corset (de jour et/ou de nuit);
- d'une activité sportive adaptée;
- d'un certificat de tiers-temps supplémentaire pour les examens, d'une dispense d'activités sportives à l'école (non recommandé mais utile dans le cas où le professeur ne s'adapte pas à l'élève handicapé par la CMT);
- de la reconnaissance d'affectation longue durée (prise en charge à 100% des frais associés aux traitements et soins en lien avec la CMT par l'Assurance Maladie);
- d'orienter les patients ayant la CMT vers des bons chirurgiens s'il y a besoin d'opérer;
- de suggérer aux patients d'ouvrir un dossier MDPH avec son médecin traitant.
En outre, le médecin MPR est très important pour une personne atteinte de la maladie de Charcot Marie-Tooth. Il est fortement recommandé d'être suivi par un médecin MPR pour suivre l'évolution de la maladie et de proposer des traitements adaptés tels que ceux sus-cités.
Classification
modifierLes différents types de maladies de Charcot-Marie-Tooth (CMT) sont classés selon trois critères[5] :
- la partie du nerf atteinte (myéline ou axone). Le critère utilisé est basé sur la vitesse de conduction de l'influx nerveux déterminée par électromyogramme. Dans le cas de l'atteinte de la myéline, cette vitesse est bien plus faible que dans le cas de l'atteinte de l'axone. Il existe également des CMT ayant une vitesse de conduction intermédiaire, classées à part ;
- le mode de transmission génétique (dominante ou récessive ou lié au chromosome X). La détermination de ce mode par le médecin résulte en grande partie d'un questionnement du patient sur l'historique de la maladie au sein de sa famille ;
- l'anomalie génétique cause de la maladie (en 2017, une soixantaine de gènes ont été identifiés).
Les deux premiers critères permettent de classer les CMT en six grands types (CMT1, CMT2, CMT4, CMTX, DI-CMT, RI-CMT), chaque type étant divisé en sous-types en fonction du troisième critère.
La variante la plus fréquente de la CMT est la CMT1A (48% des personnes atteintes), suivie de la CMT2A (6 %), la CMT1B (3 %), la CMT4C (2,5 %), la CMTX1 (2 %). Les symptômes les plus fréquents sont une instabilité des chevilles (52 %), des chutes (42 %) et une faiblesse de la main (42 %). L'atteinte est plus sévère pour la CMT2A, la CMT1B et la CMT4C que pour la CMT1A ou la CMTX1[5].
Formes démyélinisantes de la CMT
modifierDans ces types de CMT, c'est la myéline, gaine isolante entourant les nerfs, qui est atteinte. Il en résulte une baisse caractéristique de la vitesse de transmission de l'influx nerveux, souvent inférieure à 20 m/s. Ce type de CMT se traduit souvent dès la petite enfance par des difficultés en gymnastique.
On distingue :
- les CMT1, dont la transmission est autosomique dominante[6] ;
- les CMT4, dont la transmission est autosomique récessive[7].
Formes axonales de la CMT
modifierDans ces types de CMT, c'est l'axone conduisant l'influx nerveux, qui est atteint. La vitesse de conduction de l'influx nerveux n'est pas affectée (de l'ordre de 40 m/s), mais c'est l'intensité du signal électrique qui est diminuée. Elles constituent le type CMT2. On y distingue :
- les CMT2 proprement dites, dont la transmission est autosomique dominante ;
- les AR-CMT2, dont la transmission est autosomique récessive.
Formes intermédiaires de la CMT
modifierOn y regroupe des formes de CMT, où la vitesse de conduction de l'influx nerveux est comprise entre 25 m/s et 40 m/s. On distingue :
- les DI-CMT, dont la transmission est autosomique dominante ;
- les RI-CMT, dont la transmission est autosomique récessive ;
- les CMTX dont la transmission se fait par l'intermédiaire du chromosome X, soit sous forme dominante, soit sous forme récessive.
Notes et références
modifier- La maladie de Charcot-Marie-Tooth sur afm-telethon.fr
- (en) Linda Crabtree, « Charcot-Marie-Tooth Disease as a Disabling Disorder », Canadian Family Physician, vol. 35, , p. 361-367 (ISSN 0008-350X, PMID 21248895, lire en ligne [PDF], consulté le )
- (en-US) Lori A. Karol et Emily Elerson, « Scoliosis in Patients with Charcot-Marie-Tooth Disease », JBJS, vol. 89, no 7, , p. 1504 (ISSN 0021-9355, DOI 10.2106/JBJS.F.01161, lire en ligne, consulté le )
- Centre de Référence coordinateur des Maladies Neuromusculaires rares et de
la SLA - CHU La Timone, Marseille, « Synthèse à destination du médecin traitant »
[PDF], sur Myobase, (consulté le )
- AFM-Téléthon, « Avancées dans la maladie de Charcot-Marie-Tooth », sur www.afm-telethon.fr, (consulté le )
- (en) Thomas D T D Bird, Karen K Stephens, Roberta A R A Pagon, Cynthia R C R Dolan, Margaret P M P Adam (eds.), GeneReviews™, Seattle (WA), University of Washington, Seattle, (PMID 20301548, lire en ligne)
- (en) Thomas D T D Bird, Karen K Stephens, Roberta A R A Pagon, Cynthia R C R Dolan, Margaret P M P Adam (eds.), GeneReviews™, Seattle (WA), University of Washington, Seattle, (PMID 20301711, lire en ligne)
Voir aussi
modifierLiens externes
modifier- Notice dans un dictionnaire ou une encyclopédie généraliste :
- Ressources relatives à la santé :
- « CMT-France » : Association des malades de la CMT en France.
- « CMT-Québec » : Association des malades de la CMT au Québec.
- « Maladie de Charcot-Marie-Tooth », sur www.afm-france.org
- « Classification des maladies de Charcot-Marie-Tooth », sur Orphanet
- « Neuropathies héréditaires sensitivomotrice de Charcot-Marie-Tooth », sur www.has-sante.fr