Acyl group

In chemistry, an acyl group is a moiety derived by the removal of one or more hydroxyl groups from an oxoacid,[1] including inorganic acids. It contains a double-bonded oxygen atom and an organyl group (R−C=O) or hydrogen in the case of formyl group (H−C=O). In organic chemistry, the acyl group (IUPAC name alkanoyl if the organyl group is alkyl) is usually derived from a carboxylic acid, in which case it has the formula R−C(=O)−, where R represents an organyl group or hydrogen. Although the term is almost always applied to organic compounds, acyl groups can in principle be derived from other types of acids such as sulfonic acids and phosphonic acids. In the most common arrangement, acyl groups are attached to a larger molecular fragment, in which case the carbon and oxygen atoms are linked by a double bond.
Reactivity trends
[edit]There are five main types of acyl derivatives. Acid halides are the most reactive towards nucleophiles, followed by anhydrides, esters, and amides. Carboxylate ions are essentially unreactive towards nucleophilic substitution, since they possess no leaving group. The reactivity of these five classes of compounds covers a broad range; the relative reaction rates of acid chlorides and amides differ by a factor of 1013.[2]

A major factor in determining the reactivity of acyl derivatives is leaving group ability, which is related to acidity. Weak bases are better leaving groups than strong bases; a species with a strong conjugate acid (e.g. hydrochloric acid) will be a better leaving group than a species with a weak conjugate acid (e.g. acetic acid). Thus, chloride ion is a better leaving group than acetate ion. The reactivity of acyl compounds towards nucleophiles decreases as the basicity of the leaving group increases, as the table shows.[3]
| Compound Name | Structure | Leaving Group | pKa of Conjugate Acid |
|---|---|---|---|
| Acetyl chloride | 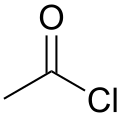 |
 |
−7 |
| Acetic anhydride |  |
 |
4.76 |
| Ethyl acetate |  |
 |
15.9 |
| Acetamide |  |
 |
38 |
| Acetate anion | |
N/a | N/a |

Another factor that plays a role in determining the reactivity of acyl compounds is resonance. Amides exhibit two main resonance forms. Both are major contributors to the overall structure, so much so that the amide bond between the carbonyl carbon and the amide nitrogen has significant double bond character. The energy barrier for rotation about an amide bond is 75–85 kJ/mol (18–20 kcal/mol), much larger than values observed for normal single bonds. For example, the C–C bond in ethane has an energy barrier of only 12 kJ/mol (3 kcal/mol).[2] Once a nucleophile attacks and a tetrahedral intermediate is formed, the energetically favorable resonance effect is lost. This helps explain why amides are one of the least reactive acyl derivatives.[3]
Esters exhibit less resonance stabilization than amides, so the formation of a tetrahedral intermediate and subsequent loss of resonance is not as energetically unfavorable. Anhydrides experience even weaker resonance stabilization, since the resonance is split between two carbonyl groups, and are more reactive than esters and amides. In acid halides, there is very little resonance, so the energetic penalty for forming a tetrahedral intermediate is small. This helps explain why acid halides are the most reactive acyl derivatives.[3]
Compounds
[edit]Well-known acyl compounds are the acyl chlorides, such as acetyl chloride (CH3COCl) and benzoyl chloride (C6H5COCl). These compounds, which are treated as sources of acylium cations, are good reagents for attaching acyl groups to various substrates. Amides (RC(O)NR′2) and esters (RC(O)OR′) are classes of acyl compounds, as are ketones (RC(O)R′) and aldehydes (RC(O)H), where R and R′ stand for organyl (or hydrogen in the case of formyl).
Acylium cations, radicals, and anions
[edit]
Acylium ions are cations of the formula RCO+.[4] The carbon–oxygen bond length in these cations is near 1.1 Å (110-112 pm), which is shorter than the 112.8 pm of carbon monoxide and indicates triple-bond character.[5][6][7]
The carbon centres of acylium ions generally have a linear geometry and sp atomic hybridization, and are best represented by a resonance structure bearing a formal positive charge on the oxygen (rather than carbon): [R−C≡O+]. They are characteristic fragments observed in EI-mass spectra of ketones.
Acylium ions are common reactive intermediates, for example in the Friedel–Crafts acylation and many other organic reactions such as the Hayashi rearrangement. Salts containing acylium ions can be generated by removal of the halide from acyl halides:
- RC(O)Cl + SbCl5 → [RCO]+[SbCl6]−
Acyl radicals are readily generated from aldehydes by hydrogen-atom abstraction. However, they undergo rapid decarbonylation to afford the alkyl radical:[8]
- RC(H)=O → RC•=O → R• + C≡O
Acyl anions are almost always unstable—usually too unstable to be exploited synthetically. They readily react with the neutral aldehyde to form an acyloin dimer. Hence, synthetic chemists have developed various acyl anion synthetic equivalents, such as dithianes, as surrogates. However, as a partial exception, hindered dialkylformamides (e.g., diisopropylformamide, HCONiPr2) can undergo deprotonation at low temperature (−78 °C) with lithium diisopropylamide as the base to form a carbamoyl anion stable at these temperatures.[9]
In biochemistry
[edit]In biochemistry there are many instances of acyl groups, in all major categories of biochemical molecules.
Acyl-CoAs are acyl derivatives formed via fatty acid metabolism. Acetyl-CoA, the most common derivative, serves as an acyl donor in many biosynthetic transformations. Such acyl compounds are thioesters.
Names of acyl groups of amino acids are formed by replacing the -ine suffix with -yl. For example, the acyl group of glycine is glycyl, and of lysine is lysyl.
Names of acyl groups of ribonucleoside monophosphates such as AMP (5′-adenylic acid), GMP (5′-guanylic acid), CMP (5′-cytidylic acid), and UMP (5′-uridylic acid) are adenylyl, guanylyl, cytidylyl, and uridylyl respectively.
In phospholipids, the acyl group of phosphatidic acid is called phosphatidyl-.
Finally, many saccharides are acylated.
In organometallic chemistry and catalysis
[edit]Acyl ligands are intermediates in many carbonylation reactions, which are important in some catalytic reactions. Metal acyls arise usually via insertion of carbon monoxide into metal–alkyl bonds. Metal acyls also arise from reactions involving acyl chlorides with low-valence metal complexes or by the reaction of organolithium compounds with metal carbonyls. Metal acyls are often described by two resonance structures, one of which emphasizes the basicity of the oxygen center. O-alkylation of metal acyls gives Fischer carbene complexes.[10]
Nomenclature
[edit]The common names of acyl groups are derived typically by replacing the -ic acid suffix of the corresponding carboxylic acid's common name with -yl (or -oyl), as shown in the table below.
In the IUPAC nomenclature of organic chemistry, the systematic names of acyl groups are derived exactly by replacing the -yl suffix of the corresponding hydrocarbyl group's systemic name (or the -oic acid suffix of the corresponding carboxylic acid's systemic name) with -oyl, as shown in the table below.
The acyls are between the hydrocarbyls and the carboxylic acids.
The hydrocarbyl group names that end in -yl are not acyl groups, but alkyl groups derived from alkanes (methyl, ethyl, propyl, butyl), alkenyl groups derived from alkenes (propenyl, butenyl), or aryl groups (benzyl).
| Corresponding hydrocarbyl group name RC– |
Acyl group name RC(O)– |
Corresponding carboxylic acid name RC(O)O-H | |||
|---|---|---|---|---|---|
| common | systematic | common | systematic | common | systematic |
| methyl | formyl | methanoyl | formic acid | methanoic acid | |
| ethyl | acetyl | ethanoyl | acetic acid | ethanoic acid | |
| propyl | propionyl | propanoyl | propionic acid | propanoic acid | |
| butyl | butyryl | butanoyl | butyric acid | butanoic acid | |
| propenyl | acrylyl or acryloyl | propenoyl | acrylic acid | propenoic acid | |
| crotyl | butenyl | crotonyl | butenoyl | crotonic acid | butenoic acid |
| benzyl | benzoyl | benzoic acid | |||
Reaction mechanisms
[edit]Acyl compounds react with nucleophiles via an addition mechanism: the nucleophile attacks the carbonyl carbon, forming a tetrahedral intermediate. This reaction can be accelerated by acidic conditions, which make the carbonyl more electrophilic, or basic conditions, which provide a more anionic and therefore more reactive nucleophile. The tetrahedral intermediate itself can be an alcohol or alkoxide, depending on the pH of the reaction.
The tetrahedral intermediate of an acyl compound contains a substituent attached to the central carbon that can act as a leaving group. After the tetrahedral intermediate forms, it collapses, recreating the carbonyl C=O bond and ejecting the leaving group in an elimination reaction. As a result of this two-step addition/elimination process, the nucleophile takes the place of the leaving group on the carbonyl compound by way of an intermediate state that does not contain a carbonyl. Both steps are reversible and as a result, nucleophilic acyl substitution reactions are equilibrium processes.[11][full citation needed] Because the equilibrium will favor the product containing the best nucleophile, the leaving group must be a comparatively poor nucleophile in order for a reaction to be practical.
Acidic conditions
[edit]Under acidic conditions, the carbonyl group of the acyl compound 1 is protonated, which activates it towards nucleophilic attack. In the second step, the protonated carbonyl 2 is attacked by a nucleophile (H−Z) to give tetrahedral intermediate 3. Proton transfer from the nucleophile (Z) to the leaving group (X) gives 4, which then collapses to eject the protonated leaving group (H−X), giving protonated carbonyl compound 5. The loss of a proton gives the substitution product, 6. Because the last step involves the loss of a proton, nucleophilic acyl substitution reactions are considered catalytic in acid. Also note that under acidic conditions, a nucleophile will typically exist in its protonated form (i.e. H−Z instead of Z−).

Basic conditions
[edit]Under basic conditions, a nucleophile (Nuc) attacks the carbonyl group of the acyl compound 1 to give tetrahedral alkoxide intermediate 2. The intermediate collapses and expels the leaving group (X) to give the substitution product 3. While nucleophilic acyl substitution reactions can be base-catalyzed, the reaction will not occur if the leaving group is a stronger base than the nucleophile (i.e. the leaving group must have a higher pKa than the nucleophile). Unlike acid-catalyzed processes, both the nucleophile and the leaving group exist as anions under basic conditions.

This mechanism is supported by isotope labeling experiments. When ethyl propionate with an oxygen-18-labeled ethoxy group is treated with sodium hydroxide (NaOH), the oxygen-18 label is completely absent from propionic acid and is found exclusively in the ethanol.[12]

Acyl species
[edit]In acyloxy groups the acyl group is bonded to oxygen: R−C(=O)−O−R′ where R−C(=O) is the acyl group.
Acylium ions are cations of the formula R−C≡O+. They are intermediates in Friedel-Crafts acylations.
See also
[edit]References
[edit]- ^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "Acyl groups". doi:10.1351/goldbook.A00123
- ^ a b Carey, Francis A. (2006). Organic Chemistry (6th ed.). New York: McGraw-Hill. pp. 866–868. ISBN 0072828374.
- ^ a b c Wade 2010, pp. 998–999.
- ^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "Acyl species". doi:10.1351/goldbook.A00129
- ^ Chevrier, B.; Carpentier, J. M. Le; Weiss, R. (1972). "Synthesis of two crystalline species of the Friedel–Crafts intermediate antimony pentachloride-p-toluoyl chloride. Crystal structures of the donor–acceptor complex and of the ionic salt". J. Am. Chem. Soc. 94 (16): 5718–5723. doi:10.1021/ja00771a031.
- ^ Davlieva, Milya G.; Lindeman, Sergey V.; Neretin, Ivan S.; Kochi, Jay K. (2004). "Structural effects of carbon monoxide coordination to carbon centers. π and σ bindings in aliphatic acyl versus aromatic aroylcations". New J. Chem. 28: 1568–1574. doi:10.1039/B407654K.
- ^ Hermannsdorfer, André; Driess, Matthias (2021). "Silicon Tetrakis(trifluoromethanesulfonate): A Simple Neutral Silane Acting as a Soft and Hard Lewis Superacid". Angew. Chem. Int. Ed. 60 (24): 13656–13660. doi:10.1002/anie.202103414. PMC 8252640. PMID 33826216.
- ^ Smith, Michael B. (2013). March's Advanced Organic Chemistry. Hoboken, NJ: Wiley. p. 857. ISBN 978-0-470-46259-1.
- ^ Fraser, Robert R.; Hubert, Patrick R. (1974-01-01). "Direct Formation of the Carbonyl Anion of Diisopropyl Formamide". Canadian Journal of Chemistry. 52 (1): 185–187. doi:10.1139/v74-029. ISSN 0008-4042.
- ^ Elschenbroich, C. (2006). Organometallics. Weinheim: Wiley-VCH. ISBN 3-527-29390-6.
- ^ Wade 2010, pp. 996–997.
- ^ McMurry, John (1996). Organic Chemistry (4th ed.). Pacific Grove, CA: Brooks/Cole Publishing Company. pp. 820–821. ISBN 0534238327.
External links
[edit]Media related to Acyl groups at Wikimedia Commons
| Authority control databases: National |
|---|