Factor VIII
| F8 | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Identifiers | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Aliases | F8, AHF, DXS1253E, F8B, F8C, FVIII, HEMA, coagulation factor VIII, THPH13 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| External IDs | OMIM: 300841; MGI: 88383; HomoloGene: 49153; GeneCards: F8; OMA:F8 - orthologs | ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||





Coagulation factor VIII (Factor VIII, FVIII, also known as anti-hemophilic factor (AHF)) is an essential blood clotting protein. In humans, it is encoded by F8 gene.[5][6] Defects in this gene result in hemophilia A, an X-linked bleeding disorder.[7]
Factor VIII is produced in the liver's sinusoidal cells and endothelial cells outside the liver throughout the body. This protein circulates in the bloodstream in an inactive form, bound to another molecule called von Willebrand factor, until an injury that damages blood vessels occurs.[8] In response to injury, coagulation factor VIII is activated and separates from von Willebrand factor. The active protein (sometimes written as coagulation factor VIIIa) interacts with another coagulation factor called factor IX. This interaction sets off a chain of additional chemical reactions that form a blood clot.[8]
Factor VIII participates in blood coagulation; it is a cofactor for factor IXa, which, in the presence of Ca2+ and phospholipids, forms a complex that converts factor X to the activated form Xa. The factor VIII gene produces two alternatively spliced transcripts. Transcript variant 1 encodes a large glycoprotein, isoform a, which circulates in plasma and associates with von Willebrand factor in a noncovalent complex. This protein undergoes multiple cleavage events. Transcript variant 2 encodes a putative small protein, isoform b, which consists primarily of the phospholipid binding domain of factor VIIIc. This binding domain is essential for coagulant activity.[9]
People with high levels of factor VIII are at increased risk for deep vein thrombosis and pulmonary embolism.[10] Copper is a required cofactor for factor VIII and copper deficiency is known to increase the activity of factor VIII.[11]
Factor VIII is available as a medication that is on the WHO Model List of Essential Medicines, the most important medications needed in a basic health system.[12]
Genetics
[edit]
Factor VIII was first characterized in 1984 by scientists at Genentech.[13] The gene for factor VIII is located on the X chromosome (Xq28). The gene for factor VIII presents an interesting primary structure, as another gene (F8A1) is embedded in one of its introns.[14]
Structure
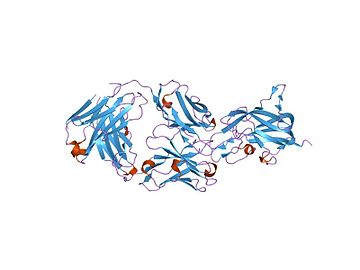
[edit]Factor VIII protein consists of six domains: A1-A2-B-A3-C1-C2, and is homologous to factor V.
The A domains are homologous to the A domains of the copper-binding protein ceruloplasmin.[15] The C domains belong to the phospholipid-binding discoidin domain family, and the C2 domain mediate membrane binding.[16]
Activation of factor VIII to factor VIIIa is done by cleavage and release of the B domain. The protein is now divided to a heavy chain, consisting of the A1-A2 domains, and a light chain, consisting of the A3-C1-C2 domains. Both form non-covalently a complex in a calcium-dependent manner. This complex is the pro-coagulant factor VIIIa.[17]
Physiology
[edit]FVIII is a glycoprotein procofactor. Although the primary site of release in humans is ambiguous, it is synthesized and released into the bloodstream by the vascular, glomerular, and tubular endothelium, and the sinusoidal cells of the liver.[18] Hemophilia A has been corrected by liver transplantation.[19] Transplanting hepatocytes was ineffective, but liver endothelial cells were effective.[19]
In the blood, it mainly circulates in a stable noncovalent complex with von Willebrand factor. Upon activation by thrombin (factor IIa), it dissociates from the complex to interact with factor IXa in the coagulation cascade. It is a cofactor to factor IXa in the activation of factor X, which, in turn, with its cofactor factor Va, activates more thrombin. Thrombin cleaves fibrinogen into fibrin which polymerizes and crosslinks (using factor XIII) into a blood clot.
The factor VIII protein has a half-life of 12 hours in the blood stream when stabilized by the von Willebrand factor. [20]
No longer protected by vWF, activated FVIII is proteolytically inactivated in the process (most prominently by activated protein C and factor IXa) and quickly cleared from the blood stream.
Factor VIII is not affected by liver disease. In fact, levels usually are elevated in such instances.[21][22]
Medical use
[edit]
FVIII concentrated from donated blood plasma, or recombinant FVIII can be given to hemophiliacs to restore hemostasis. Bypassing agents such as recombinant FVIIa can be used in acquired hemophilia.
Antibody formation to factor VIII can also be a major concern for patients receiving therapy against bleeding; the incidence of these inhibitors is dependent of various factors, including the factor VIII product itself.[23]
Immunostain target
[edit]Factor VIII related antigen is used as a target for immunohistochemistry, where endothelial cells, megakaryocytes, platelets and mast cells normally stain positive.[24]
Contamination scandal
[edit]In the 1980s, some pharmaceutical companies such as Baxter International and Bayer sparked controversy by continuing to sell contaminated factor VIII after new heat-treated versions were available.[25] Under FDA pressure, unheated product was pulled from US markets, but was sold to Asian, Latin American, and some European countries. The product was tainted with HIV, a concern that had been discussed by Bayer and the U.S. Food and Drug Administration (FDA).[25]
In the early 1990s, pharmaceutical companies began to produce recombinant synthesized factor products, which now prevent nearly all forms of disease transmission during replacement therapy.
History
[edit]Factor VIII was first discovered in 1937, but it was not until 1979 that its purification by Edward Tuddenham, Frances Rotblat and coworkers led to the molecular identification of the protein.[26][27]
See also
[edit]References
[edit]- ^ a b c GRCh38: Ensembl release 89: ENSG00000185010 – Ensembl, May 2017
- ^ a b c GRCm38: Ensembl release 89: ENSMUSG00000031196 – Ensembl, May 2017
- ^ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ Toole JJ, Knopf JL, Wozney JM, Sultzman LA, Buecker JL, Pittman DD, et al. (1984). "Molecular cloning of a cDNA encoding human antihaemophilic factor". Nature. 312 (5992): 342–347. Bibcode:1984Natur.312..342T. doi:10.1038/312342a0. PMID 6438528. S2CID 4313575.
- ^ Truett MA, Blacher R, Burke RL, Caput D, Chu C, Dina D, et al. (October 1985). "Characterization of the polypeptide composition of human factor VIII:C and the nucleotide sequence and expression of the human kidney cDNA". DNA. 4 (5): 333–349. doi:10.1089/dna.1985.4.333. PMID 3935400.
- ^ Antonarakis SE (July 1995). "Molecular genetics of coagulation factor VIII gene and hemophilia A". Thrombosis and Haemostasis. 74 (1): 322–328. doi:10.1055/s-0038-1642697. PMID 8578479. S2CID 23435953.
- ^ a b "NIH: F8 – coagulation factor VIII". National Institutes of Health.
- ^ "Entrez Gene: F8 coagulation factor VIII, procoagulant component (hemophilia A)".
- ^ Jenkins PV, Rawley O, Smith OP, O'Donnell JS (June 2012). "Elevated factor VIII levels and risk of venous thrombosis". British Journal of Haematology. 157 (6): 653–663. doi:10.1111/j.1365-2141.2012.09134.x. PMID 22530883.
- ^ Milne DB, Nielsen FH (March 1996). "Effects of a diet low in copper on copper-status indicators in postmenopausal women". The American Journal of Clinical Nutrition. 63 (3): 358–364. doi:10.1093/ajcn/63.3.358. PMID 8602593.
- ^ "19th WHO Model List of Essential Medicines" (PDF). WHO. April 2015. Retrieved May 10, 2015.
- ^ Gitschier J, Wood WI, Goralka TM, Wion KL, Chen EY, Eaton DH, et al. (November 1984). "Characterization of the human factor VIII gene". Nature. 312 (5992): 326–330. Bibcode:1984Natur.312..326G. doi:10.1038/312326a0. PMID 6438525. S2CID 4358041.
- ^ Levinson B, Kenwrick S, Lakich D, Hammonds G, Gitschier J (May 1990). "A transcribed gene in an intron of the human factor VIII gene". Genomics. 7 (1): 1–11. doi:10.1016/0888-7543(90)90512-S. PMID 2110545.
- ^ Villoutreix BO, Dahlbäck B (June 1998). "Structural investigation of the A domains of human blood coagulation factor V by molecular modeling". Protein Science. 7 (6): 1317–1325. doi:10.1002/pro.5560070607. PMC 2144041. PMID 9655335.
- ^ Macedo-Ribeiro S, Bode W, Huber R, Quinn-Allen MA, Kim SW, Ortel TL, et al. (November 1999). "Crystal structures of the membrane-binding C2 domain of human coagulation factor V". Nature. 402 (6760): 434–439. Bibcode:1999Natur.402..434M. doi:10.1038/46594. PMID 10586886. S2CID 4393638.
- ^ Thorelli E, Kaufman RJ, Dahlbäck B (June 1998). "The C-terminal region of the factor V B-domain is crucial for the anticoagulant activity of factor V". The Journal of Biological Chemistry. 273 (26): 16140–16145. doi:10.1074/jbc.273.26.16140. PMID 9632668.
- ^ Kumar V, Abbas A, Aster J (2005). Robbins and Cotran Pathologic Basis of Disease (9th ed.). Pennsylvania: Elsevier. p. 655. ISBN 978-0-8089-2450-0.
- ^ a b Kaushansky K, Lichtman M, Beutler E, Kipps T, Prchal J, Seligsohn U (2010). Williams Hematology (8th ed.). McGraw-Hill. ISBN 978-0-07-162151-9.
- ^ Fischer K, Pendu R, van Schooten CJ, van Dijk K, Denis CV, van den Berg HM, et al. (August 2009). "Models for prediction of factor VIII half-life in severe haemophiliacs: distinct approaches for blood group O and non-O patients". PLOS ONE. 4 (8): e6745. Bibcode:2009PLoSO...4.6745F. doi:10.1371/journal.pone.0006745. PMC 2727052. PMID 19707594.
- ^ Hollestelle MJ, Geertzen HG, Straatsburg IH, van Gulik TM, van Mourik JA (February 2004). "Factor VIII expression in liver disease". Thrombosis and Haemostasis. 91 (2): 267–275. doi:10.1160/th03-05-0310. PMID 14961153. S2CID 20091477.
- ^ Rubin R, Leopold L (1998). Hematologic Pathophysiology. Madison, Conn: Fence Creek Publishing. ISBN 1-889325-04-X.
- ^ Lozier J (2004). "Overview of Factor VIII Inhibitors". CMEonHemophilia.com. Archived from the original on 2008-12-16. Retrieved 2009-01-07.
- ^ Pernick N. "Stains & CD markers - Factor VIII related antigen". Pathology Outlines. Topic Completed: 1 July 2012. Minor changes: 25 June 2021
- ^ a b Bogdanich W, Koli E (May 2003). "2 paths of Bayer drug in 80's: riskier one steered overseas". The New York Times on the Web: A1, C5. PMID 12812170. Retrieved 2009-01-07.
- ^ Tuddenham EG, Trabold NC, Collins JA, Hoyer LW (January 1979). "The properties of factor VIII coagulant activity prepared by immunoadsorbent chromatography". The Journal of Laboratory and Clinical Medicine. 93 (1): 40–53. PMID 366050.
- ^ "Frances Rotblat obituary". The Times. 12 June 2021. Retrieved 12 June 2021.
Further reading
[edit]- Gitschier J (1991). "The molecular basis of hemophilia A". Annals of the New York Academy of Sciences. 614 (1 Process in Va): 89–96. Bibcode:1991NYASA.614...89G. doi:10.1111/j.1749-6632.1991.tb43694.x. PMID 1902642. S2CID 26493612.
- White GC, Shoemaker CB (January 1989). "Factor VIII gene and hemophilia A". Blood. 73 (1): 1–12. PMID 2491949.
- Antonarakis SE, Kazazian HH, Tuddenham EG (1995). "Molecular etiology of factor VIII deficiency in hemophilia A". Human Mutation. 5 (1): 1–22. doi:10.1002/humu.1380050102. PMID 7728145. S2CID 2346510.
- Lenting PJ, van Mourik JA, Mertens K (December 1998). "The life cycle of coagulation factor VIII in view of its structure and function". Blood. 92 (11): 3983–3996. doi:10.1182/blood.V92.11.3983. PMID 9834200.
- Saenko EL, Ananyeva N, Kouiavskaia D, Schwinn H, Josic D, Shima M, et al. (August 2002). "Molecular defects in coagulation Factor VIII and their impact on Factor VIII function". Vox Sanguinis. 83 (2): 89–96. doi:10.1046/j.1423-0410.2002.00183.x. hdl:2027.42/74861. PMID 12201837.
- Lollar P (August 2002). "Molecular characterization of the immune response to factor VIII". Vox Sanguinis. 83. 83 (Suppl 1): 403–408. doi:10.1111/j.1423-0410.2002.tb05342.x. PMID 12617176. S2CID 26050729.
- Fay PJ (March 2004). "Activation of factor VIII and mechanisms of cofactor action". Blood Reviews. 18 (1): 1–15. doi:10.1016/S0268-960X(03)00025-0. PMID 14684146.
- Lavigne-Lissalde G, Schved JF, Granier C, Villard S (October 2005). "Anti-factor VIII antibodies: a 2005 update". Thrombosis and Haemostasis. 94 (4): 760–769. doi:10.1160/TH05-02-0118. PMID 16270627. S2CID 38533008.
- Fang H, Wang L, Wang H (2007). "The protein structure and effect of factor VIII". Thrombosis Research. 119 (1): 1–13. doi:10.1016/j.thromres.2005.12.015. PMID 16487577.
External links
[edit]- GeneReviews/NCBI/NIH/UW entry on Hemophilia A
- The Coagulation Factor VIII Protein
- Factor+VIII at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- Overview of all the structural information available in the PDB for UniProt: P00451 (Human Factor VIII) at the PDBe-KB.
PDB gallery | |
|---|---|
|