Calcium channel blocker
| Calcium channel blockers | |
|---|---|
| Drug class | |
| Class identifiers | |
| Use | hypertension, arrhythmia, cluster headache[1] |
| ATC code | C08 |
| External links | |
| MeSH | D002121 |
| Legal status | |
| In Wikidata | |
Calcium channel blockers (CCB), calcium channel antagonists or calcium antagonists[2] are a group of medications that disrupt the movement of calcium (Ca2+
) through calcium channels.[3] Calcium channel blockers are used as antihypertensive drugs, i.e., as medications to decrease blood pressure in patients with hypertension. CCBs are particularly effective against large vessel stiffness, one of the common causes of elevated systolic blood pressure in elderly patients.[4] Calcium channel blockers are also frequently used to alter heart rate (especially from atrial fibrillation), to prevent peripheral and cerebral vasospasm, and to reduce chest pain caused by angina pectoris.
N-type, L-type, and T-type voltage-dependent calcium channels are present in the zona glomerulosa of the human adrenal gland, and CCBs can directly influence the biosynthesis of aldosterone in adrenocortical cells, with consequent impact on the clinical treatment of hypertension with these agents.[5]
CCBs have been shown to be slightly more effective than beta blockers at lowering cardiovascular mortality associated with stroke, but they are associated with more side effects.[6][7] Potential major risks however were mainly found to be associated with short-acting CCBs.[8]
Classes
[edit]Dihydropyridine
[edit]
Dihydropyridine (DHP) calcium channel blockers are derived from the molecule dihydropyridine and often used to reduce systemic vascular resistance and arterial pressure. Sometimes when they are used to treat angina, the vasodilation and hypotension can lead to reflex tachycardia, which can be detrimental for patients with ischemic symptoms because of the resulting increase in myocardial oxygen demand. Dihydropyridine calcium channel blockers can worsen proteinuria in patients with nephropathy.[9]
This CCB class is easily identified by the suffix "-dipine".
- Amlodipine (Norvasc)
- Aranidipine (Sapresta)
- Azelnidipine (Calblock)
- Barnidipine (HypoCa)
- Benidipine (Coniel)
- Cilnidipine (Atelec, Cinalong, Siscard) Not available in US
- Clevidipine (Cleviprex)
- Efonidipine (Landel)
- Felodipine (Plendil)
- Isradipine (DynaCirc, Prescal)
- Lacidipine (Motens, Lacipil)
- Lercanidipine (Zanidip)
- Manidipine (Calslot, Madipine)
- Nicardipine (Cardene, Carden SR)
- Nifedipine (Procardia, Adalat)
- Nilvadipine (Nivadil)
- Nimodipine (Nimotop) This substance can pass the blood-brain barrier and is used to prevent cerebral vasospasm.
- Nisoldipine (Baymycard, Sular, Syscor)
- Nitrendipine (Cardif, Nitrepin, Baylotensin)
- Pranidipine (Acalas)
Non-dihydropyridine
[edit]Phenylalkylamine
[edit]
Phenylalkylamine calcium channel blockers are relatively selective for myocardium, reduce myocardial oxygen demand and reverse coronary vasospasm, and are often used to treat angina. They have minimal vasodilatory effects compared with dihydropyridines and therefore cause less reflex tachycardia, making it appealing for treatment of angina, where tachycardia can be the most significant contributor to the heart's need for oxygen. Therefore, as vasodilation is minimal with the phenylalkylamines, the major mechanism of action is causing negative inotropy. Phenylalkylamines are thought to access calcium channels from the intracellular side, although the evidence is somewhat mixed.[10]
- Fendiline
- Gallopamil
- Verapamil (Calan, Isoptin)
Benzothiazepine
[edit]
Benzothiazepine calcium channel blockers belong to the benzothiazepine class of compounds and are an intermediate class between phenylalkylamine and dihydropyridines in their selectivity for vascular calcium channels. By having both cardiac depressant and vasodilator actions, benzothiazepines are able to reduce arterial pressure without producing the same degree of reflex cardiac stimulation caused by dihydropyridines.
- Diltiazem (Cardizem) (also used experimentally to prevent migraine)[citation needed]
Nonselective
[edit]While most of the agents listed above are relatively selective, there are additional agents that are considered nonselective. These include mibefradil, bepridil, flunarizine (BBB crossing), fluspirilene (BBB crossing),[11] and fendiline.[12]
Others
[edit]Gabapentinoids, such as gabapentin and pregabalin, are selective blockers of α2δ subunit-containing voltage-gated calcium channels. They are used primarily to treat epilepsy and neuropathic pain.[13]
Ziconotide, a peptide compound derived from the omega-conotoxin, is a selective N-type calcium channel blocker that has potent analgesic properties that are equivalent to approximate 1,000 times that of morphine. It must be delivered via the intrathecal (directly into the cerebrospinal fluid) route via an intrathecal infusion pump.[14]
Naturally occurring compounds and elements such as magnesium have also been shown to act as calcium channel blockers when administered orally.[15]
Side effects
[edit]Side effects of these drugs may include but are not limited to:
- Constipation
- Peripheral edema, which can occur in as much as 70% of people receiving calcium channel blocker, is caused by calcium channel blockers' preferential arteriolar or precapillary dilation without commensurate dilation in the venous or postcapillary circulation.[16][17][18][19][20] Since lymphatic drainage relies on contraction of the smooth muscle inside the lymphatic vessel[21] supported by voltage-gated calcium channels, inhibition of voltage-gated calcium channel poses a threat towards lymphatic removal of interstitial fluid essential for normal lymphatic system functioning.[20] (See also: Lymphedema.)
- Gingival overgrowth
Toxicity
[edit]
Mild CCB toxicity is treated with supportive care. Nondihydropyridine CCBs may produce profound toxicity, and early decontamination, especially for slow-release agents, is essential. For severe overdoses, treatment usually includes close monitoring of vital signs and the addition of vasopressive agents and intravenous fluids for blood pressure support. Intravenous calcium gluconate (or calcium chloride if a central line is available) and atropine are first-line therapies. If the time of the overdose is known and presentation is within two hours of ingestion, activated charcoal, gastric lavage, and polyethylene glycol may be used to decontaminate the gut. Efforts for gut decontamination may be extended to within 8 hours of ingestion with extended-release preparations.[citation needed]
Hyperinsulinemia-euglycemia therapy has emerged as a viable form of treatment.[22] Although the mechanism is unclear, increased insulin may mobilize glucose from peripheral tissues to serve as an alternative fuel source for the heart (the heart mainly relies on oxidation of fatty acids). Theoretical treatment with lipid emulsion therapy has been considered in severe cases, but is not yet standard of care.
Caution should be taken when using verapamil with a beta blocker due to the risk of severe bradycardia. If unsuccessful, ventricular pacing should be used.[23]
Non-medical calcium channel inhibitors
[edit]Ethanol
[edit]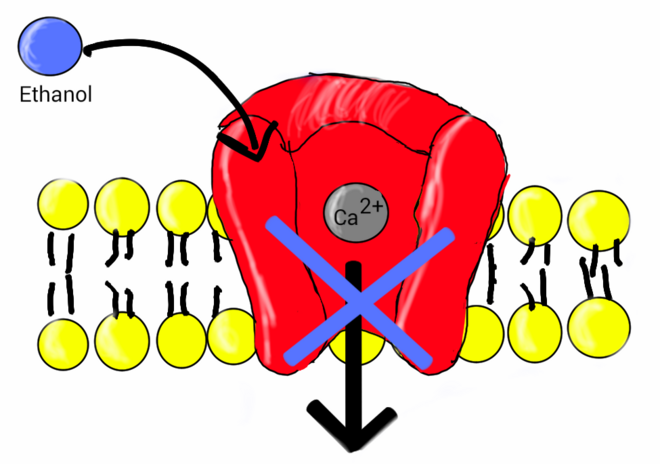
Research indicates ethanol is involved in the inhibition of L-type calcium channels. One study showed the nature of ethanol binding to L-type calcium channels is according to first-order kinetics with a Hill coefficient around 1. This indicates ethanol binds independently to the channel, expressing noncooperative binding.[24] Early studies showed a link between calcium and the release of vasopressin by the secondary messenger system.[25] Vasopressin levels are reduced after the ingestion of alcohol.[26] The lower levels of vasopressin from the consumption of alcohol have been linked to ethanol acting as an antagonist to voltage-gated calcium channels (VGCCs). Studies conducted by Treistman et al. in the aplysia confirm inhibition of VGCC by ethanol. Voltage clamp recordings have been done on the aplysia neuron. VGCCs were isolated and calcium current was recorded using patch clamp technique having ethanol as a treatment. Recordings were replicated at varying concentrations (0, 10, 25, 50, and 100 mM) at a voltage clamp of +30 mV. Results showed calcium current decreased as concentration of ethanol increased.[27] Similar results have shown to be true in single-channel recordings from isolated nerve terminal of rats that ethanol does in fact block VGCCs.[28]
Studies done by Katsura et al. in 2006 on mouse cerebral cortical neurons, show the effects of prolonged ethanol exposure. Neurons were exposed to sustained ethanol concentrations of 50 mM for 3 days in vitro. Western blot and protein analysis were conducted to determine the relative amounts of VGCC subunit expression. α1C, α1D, and α2/δ1 subunits showed an increase of expression after sustained ethanol exposure. However, the β4 subunit showed a decrease. Furthermore, α1A, α1B, and α1F subunits did not alter in their relative expression. Thus, sustained ethanol exposure may participate in the development of ethanol dependence in neurons.[29]
Other experiments done by Malysz et al. have looked into ethanol effects on voltage-gated calcium channels on detrusor smooth muscle cells in guinea pigs. Perforated patch clamp technique was used having intracellular fluid inside the pipette and extracellular fluid in the bath with added 0.3% vol/vol (about 50-mM) ethanol. Ethanol decreased the Ca2+
current in DSM cells and induced muscle relaxation. Ethanol inhibits VGCCs and is involved in alcohol-induced relaxation of the urinary bladder.[30]
Agatoxin in spider venom
[edit]Research on the desert grass spider, Agelenopsis aperta, has shown that agatoxins IVA and IVB found in their venom selectively block calcium channels. These agatoxins are found in other spider species as well. Desert grass spider bites to insects result in rapid paralysis, but bites to humans are not considered medically significant.[31]
Mechanism of action
[edit]
In the body's tissues, the concentration of calcium ions (Ca2+
) outside cells is normally about 10,000-fold higher than the concentration inside cells. Embedded in the membrane of some cells are calcium channels. When these cells receive a certain signal, the channels open, letting calcium rush into the cell. The resulting increase in intracellular calcium has different effects in different types of cells. Calcium channel blockers prevent or reduce the opening of these channels and thereby reduce these effects.[citation needed]
Several types of calcium channels occur, with a number of classes of blockers, but almost all of them preferentially or exclusively block the L-type voltage-gated calcium channel.[32]
Voltage-dependent calcium channels are responsible for excitation-contraction coupling of skeletal, smooth, and cardiac muscle and for regulating aldosterone and cortisol secretion in endocrine cells of the adrenal cortex.[5] In the heart, they are also involved in the conduction of the pacemaker signals. CCBs used as medications primarily have four effects:
- By acting on vascular smooth muscle, they reduce contraction of the arteries and cause an increase in arterial diameter, a phenomenon called vasodilation (CCBs do not work on venous smooth muscle).
- By acting on cardiac muscles (myocardium), they reduce the force of contraction of the heart.
- By slowing down the conduction of electrical activity within the heart, they slow down the heart beat.
- By blocking the calcium signal on adrenal cortex cells, they directly reduce aldosterone production, which correlates to lower blood pressure.
Since blood pressure is in intimate feedback with cardiac output and peripheral resistance, with relatively low blood pressure, the afterload on the heart decreases; this decreases how hard the heart must work to eject blood into the aorta, so the amount of oxygen required by the heart decreases accordingly. This can help ameliorate symptoms of ischaemic heart disease such as angina pectoris.

Reducing the force of contraction of the myocardium is known as the negative inotropic effect of calcium channel blockers.
Slowing down the conduction of electrical activity within the heart, by blocking the calcium channel during the plateau phase of the action potential of the heart (see: cardiac action potential), results in a negative chronotropic effect, or a lowering of heart rate. This can increase the potential for heart block. The negative chronotropic effects of CCBs make them a commonly used class of agents in individuals with atrial fibrillation or flutter in whom control of the heart rate is generally a goal. Negative chronotropy can be beneficial when treating a variety of disease processes because lower heart rates represent lower cardiac oxygen requirements. Elevated heart rate can result in significantly higher "cardiac work", which can result in symptoms of angina.
The class of CCBs known as dihydropyridines mainly affect arterial vascular smooth muscle and lower blood pressure by causing vasodilation. The phenylalkylamine class of CCBs mainly affect the cells of the heart and have negative inotropic and negative chronotropic effects. The benzothiazepine class of CCBs combine effects of the other two classes.
Because of the negative inotropic effects, the nondihydropyridine calcium channel blockers should be avoided (or used with caution) in individuals with cardiomyopathy.[33]
Unlike beta blockers, calcium channel blockers do not decrease the responsiveness of the heart to input from the sympathetic nervous system. Since moment-to-moment blood pressure regulation is carried out by the sympathetic nervous system (via the baroreceptor reflex), calcium channel blockers allow blood pressure to be maintained more effectively than do beta blockers. However, because dihydropyridine CCBs result in a decrease in blood pressure, the baroreceptor reflex often initiates a reflexive increase in sympathetic activity leading to increased heart rate and contractility.
Ionic calcium is antagonized by magnesium ions in the nervous system. Because of this, bioavailable supplements of magnesium, possibly including magnesium chloride, magnesium lactate, and magnesium aspartate, may increase or enhance the effects of calcium channel blockade.[34]
N-type calcium channels are found in neurons and are involved in the release of neurotransmitter at synapses. Ziconotide is a selective blocker of these calcium channels and acts as an analgesic.[14]
History
[edit]Calcium channel blockers came into wide use in the 1960s,[35] having been first identified in the lab of German pharmacologist Albrecht Fleckenstein in 1964.[36]
References
[edit]- ^ Tfelt-Hansen P, Tfelt-Hansen J (2009). "Verapamil for cluster headache. Clinical pharmacology and possible mode of action". Headache: The Journal of Head and Face Pain. 49 (1): 117–25. doi:10.1111/j.1526-4610.2008.01298.x. PMID 19125880.
- ^ Olson K (2011). "40. Calcium Channel Antagonists". Poisoning & drug overdose (6th ed.). McGraw-Hill Medical. ISBN 978-0-07-166833-0.
- ^ "calcium channel blocker" at Dorland's Medical Dictionary
- ^ Nelson M (2010). "Drug treatment of elevated blood pressure". Australian Prescriber. 33 (4): 108–12. doi:10.18773/austprescr.2010.055.
- ^ a b c Felizola SJ, Maekawa T, Nakamura Y, Satoh F, Ono Y, Kikuchi K, Aritomi S, Ikeda K, Yoshimura M, Tojo K, Sasano H (2014). "Voltage-gated calcium channels in the human adrenal and primary aldosteronism". J Steroid Biochem Mol Biol. 144 (part B): 410–16. doi:10.1016/j.jsbmb.2014.08.012. PMID 25151951. S2CID 23622821.
- ^ Chen N, Zhou M, Yang M, Guo J, Zhu C, Yang J, Wang Y, Yang X, He L (2010). "Calcium channel blockers versus other classes of drugs for hypertension". Cochrane Database of Systematic Reviews. 8 (8): CD003654. doi:10.1002/14651858.CD003654.pub4. PMID 20687074.
- ^ "Calcium Channel Blockers". MedicineNet. p. 2. Archived from the original on 2012-04-21. Retrieved 2013-01-19.
- ^ Norman M Kaplan, MD, Burton D Rose, MD (Apr 3, 2000). "Major side effects and safety of calcium channel blockers". Chinese Medical & Biological Information. Archived from the original on December 30, 2011. Retrieved July 23, 2012.
- ^ Remuzzi G, Scheppati A, Ruggenenti P (2002). "Clinical Practice. Nephropathy in Patients with Type 2 Diabetes". New England Journal of Medicine. 346 (15): 1145–51. doi:10.1056/NEJMcp011773. PMID 11948275.
- ^ Hockerman, G.H., Peterson, B.Z., Johnson, B.D., Catterall, W.A. (1997). "Molecular Determinants of Drug Binding and Action on L-Type Calcium Channels". Annual Review of Pharmacology and Toxicology. 37: 361–96. doi:10.1146/annurev.pharmtox.37.1.361. PMID 9131258. S2CID 16275155.
- ^ Bezprozvanny I, Tsien RW (1995). "Voltage-Dependent Blockade of Diverse Types of Voltage-Gated Ca2+
Channels Expressed in Xenopus Oocytes by the Ca2+
Channel Antagonist Mibefradil (Ro 40-5967)". Molecular Pharmacology. 48 (3): 540–49. PMID 7565636. - ^ Scultéty S, Tamáskovits E (1991). "Effect of Ca2+
Antagonists on Isolated Rabbit Detrusor Muscle". Acta Physiologica Hungarica. 77 (3–4): 269–78. PMID 1755331. - ^ Zamponi GW, Striessnig J, Koschak A, Dolphin AC (October 2015). "The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential". Pharmacological Reviews. 67 (4): 821–70. doi:10.1124/pr.114.009654. PMC 4630564. PMID 26362469.
- ^ a b McDowell GC, Pope JE (July 2016). "Intrathecal Ziconotide: Dosing and Administration Strategies in Patients With Refractory Chronic Pain". Neuromodulation. 19 (5): 522–32. doi:10.1111/ner.12392. PMC 5067570. PMID 26856969.
- ^ Houston M (2011). "The role of magnesium in hypertension and cardiovascular disease". Journal of Clinical Hypertension (Greenwich, Conn.). 13 (11): 843–847. doi:10.1111/j.1751-7176.2011.00538.x. PMC 8108907. PMID 22051430.
- ^ Sica DA (2003). "Calcium Channel Blocker-Related Peripheral Edema: Can It Be Resolved?". The Journal of Clinical Hypertension. 5 (4). Wiley: 291–295. doi:10.1111/j.1524-6175.2003.02402.x. ISSN 1524-6175. PMC 8099365. PMID 12939574.
- ^ "Calcium-Channel Blockers (CCBs)". CV Pharmacology. Retrieved 2020-02-07.
- ^ Domenic A. Sica. "Calcium Channel Blocker-Related Peripheral Edema". Medscape. Retrieved 2019-10-26.
- ^ Matthew R. Weir. "Incidence of Pedal Edema Formation With Dihydropyridine Calcium". Medscape. Retrieved 2019-10-26.
- ^ a b Mohanakumar S, Telinius N, Kelly B, Hjortdal V (2019-08-20). "Reduced Lymphatic Function Predisposes to Calcium Channel Blocker Edema: A Randomized Placebo-Controlled Clinical Trial". Lymphatic Research and Biology. 18 (2). Mary Ann Liebert Inc: 156–165. doi:10.1089/lrb.2019.0028. ISSN 1539-6851. PMID 31429625. S2CID 201094829.
- ^ Babak Mehrara. John F Eidt, Joseph L Mills Sr, Harold J Burstein, Kathryn A Collins (eds.). "Clinical features and diagnosis of peripheral lymphedema". UpToDate. Retrieved 2019-10-27.
- ^ Engebretsen KM, Kaczmarek KM, Morgan J, Holger JS (2011). "High-dose insulin therapy in beta-blocker and calcium channel-blocker poisoning". Clinical Toxicology. 49 (4): 277–283. doi:10.3109/15563650.2011.582471. ISSN 1556-9519. PMID 21563902. S2CID 32138463.
- ^ Buckley N, Dawson A, Whyte I (2007). "Calcium Channel Blockers". Medicine. 35 (11): 599–602. doi:10.1016/j.mpmed.2007.08.025.
- ^ Wang X, Wang G, Lemos JR, Treistman SN (September 1994). "Ethanol directly modulates gating of a dihydropyridine-sensitive Ca2+
channel in neurohypophysial terminals". J. Neurosci. 14 (9): 5453–60. doi:10.1523/JNEUROSCI.14-09-05453.1994. PMC 6577079. PMID 7521910. - ^ Tobin V, Leng G, Ludwig M (2012). "The involvement of actin, calcium channels and exocytosis proteins in somato-dendritic oxytocin and vasopressin release". Front Physiol. 3: 261. doi:10.3389/fphys.2012.00261. PMC 3429037. PMID 22934017.
- ^ Chiodera P, Coiro V (May 1990). "Inhibitory effect of ethanol on the arginine vasopressin response to insulin-induced hypoglycemia and the role of endogenous opioids". Neuroendocrinology. 51 (5): 501–04. doi:10.1159/000125383. PMID 2112727.
- ^ Treistman SN, Bayley H, Lemos JR, Wang XM, Nordmann JJ, Grant AJ (1991). "Effects of ethanol on calcium channels, potassium channels, and vasopressin release". Ann. N. Y. Acad. Sci. 625 (1): 249–63. Bibcode:1991NYASA.625..249T. doi:10.1111/j.1749-6632.1991.tb33844.x. PMID 1647726. S2CID 28281696.
- ^ Walter HJ, Messing RO (August 1999). "Regulation of neuronal voltage-gated calcium channels by ethanol". Neurochem. Int. 35 (2): 95–101. doi:10.1016/s0197-0186(99)00050-9. PMID 10405992. S2CID 36172178.
- ^ Katsura M, Shibasaki M, Hayashida S, Torigoe F, Tsujimura A, Ohkuma S (October 2006). "Increase in expression of α1 and α2/δ1 subunits of L-type high voltage-gated calcium channels after sustained ethanol exposure in cerebral cortical neurons". J. Pharmacol. Sci. 102 (2): 221–30. doi:10.1254/jphs.fp0060781. PMID 17031067.
- ^ Malysz J, Afeli SA, Provence A, Petkov GV (January 2014). "Ethanol-mediated relaxation of guinea pig urinary bladder smooth muscle: involvement of BK and L-type Ca2+
channels". Am. J. Physiol., Cell Physiol. 306 (1): C45–58. doi:10.1152/ajpcell.00047.2013. PMC 3919972. PMID 24153429. - ^ Adams ME (April 2004). "Agatoxins: ion channel specific toxins from the american funnel web spider, Agelenopsis aperta". Toxicon. 43 (5): 509–525. Bibcode:2004Txcn...43..509A. doi:10.1016/j.toxicon.2004.02.004. ISSN 0041-0101. PMID 15066410.
- ^ Yousef, et al. (2005). "The mechanism of action of calcium channel blockers in the treatment of diabetic nephropathy" (PDF). Int J Diabetes & Metabolism. 13 (2): 76–82. doi:10.1159/000497574. Archived from the original (PDF) on 2015-10-10. Retrieved 2013-06-29.
- ^ Lehne R (2010). Pharmacology for Nursing Care (7th ed.). St. Louis, Missouri: Saunders Elsevier. p. 505. ISBN 978-1-4160-6249-3.
- ^ Iseri LT, French JH (1984). "Magnesium: Nature's Physiologic Calcium Blocker". American Heart Journal. 108 (1): 188–93. doi:10.1016/0002-8703(84)90572-6. PMID 6375330.
- ^ Tekol, Y. (2007). "The medieval physician Avicenna used an herbal calcium channel blocker, Taxus baccata L". Phytotherapy Research. 21 (7): 701–02. doi:10.1002/ptr.2173. PMID 17533639. S2CID 42060942.
- ^ Fleckenstein A (1983). "History of calcium antagonists". Circulation Research. 52 (2 Pt 2): 13–16. PMID 6339106.
External links
[edit]- Calcium+Channel+Blockers at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- "Official Adalat (Nifedipine) site". Bayer. Archived from the original on 2008-04-08. Retrieved 2021-06-18.
- Video – Calcium Channel Blockers
Major chemical drug groups – based upon the Anatomical Therapeutic Chemical Classification System | |
|---|---|
| gastrointestinal tract / metabolism (A) | |
| blood and blood forming organs (B) | |
| cardiovascular system (C) | |
| skin (D) | |
| genitourinary system (G) | |
| endocrine system (H) | |
| infections and infestations (J, P, QI) | |
| malignant disease (L01–L02) | |
| immune disease (L03–L04) | |
| muscles, bones, and joints (M) | |
| brain and nervous system (N) |
|
| respiratory system (R) | |
| sensory organs (S) | |
| other ATC (V) | |