Arbre filogenètic


Un arbre filogenètic és un arbre que mostra les relacions evolutives entre diverses espècies o altres entitats que es creu que van tenir una descendència comuna, com per exemple per les llengües.
En biologia s'utilitzen aquests gràfics per a conèixer com es troben emparentats els organismes vius, de manera que cada branca representa un descendent de l'antecessor representat pel node. Cada un dels nodes s'anomena unitat taxonòmica.
A diferència dels arbres genealògics, en els quals s'utilitza informació proporcionada pels familiars, per als arbres filogenètics s'usa informació provinent de fòssils així com aquella generada per la comparança estructural i molecular dels organismes. Cal recordar que en els arbres filogenètics es mostren les relacions entre espècies i no entre individus com és el cas dels genealògics.
Els arbres filogenètics es construeixen tenint en compte la teoria de l'evolució, que indica que tots els organismes són descendents d'un ancestre comú: la protocèl·lula. Així, tots els organismes, ja siguin vius o extints, es troben emparentats en algun grau.
No obstant això, els arbres filogenètics no consideren les relacions horitzontals, per això s'estan desenvolupant altres representacions del tipus xarxa filogenètica.
Història
[modifica]
La noció d'arbre de la vida no és pas nova, ja des de l'antiguitat s'han descrit escales de progressió des de les formes més baixes a les més altes (un exemple seria la gran cadena dels éssers de Charles Darwin (1859) que va il·lustrar i popularitzar per primera vegada la noció d'arbre evolutiu en el seu llibre L'origen de les espècies). Un segle després la biologia evolutiva encara usa diagrames en forma d'arbre per descriure l'evolució.
Tipus d'arbres filogenètics
[modifica]
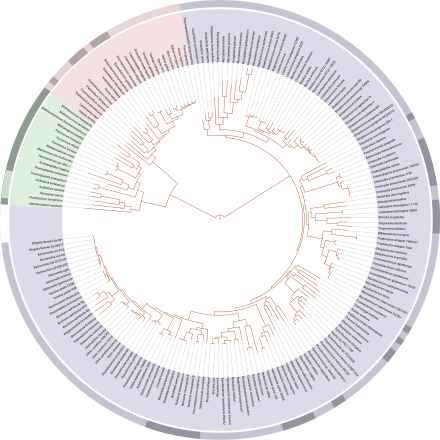

Un arbre filogenètic arrelat és un arbre directe, amb un únic nòdul que correspon a l'ancestre comú més recent de totes les entitats de les fulles de l'arbre. La Figura 1 descriu un arbre filogenètic arrelat acolorit d'acord amb el "sistema de tres dominis".[4] El mètode més comú per a arrelar els arbres és l'ús d'un grup extern no controvertit i prou proper per a permetre la inferència de les dades de seqüència o trets, però prou llunyà per a ser un grup extern evident.
Els arbres genètics sense arrel il·lustren la relació dels nòduls de les fulles sense fer assumpcions sobre ascendència. Mentre que els arbres sense arrel sempre poden ser generats a partir d'arbres arrelats ometent l'arrel, una arrel no pot ser inferida a partir d'un arbre sense arrel sense alguns mitjans per a identificar l'ascendència. Això sol fer-se incloent un grup extern en les dades entrants o introduint assumpcions addicionals sobre les taxes relatives d'evolució en cada branca, com una aplicació de la hipòtesi del rellotge molecular. La Figura 2 descriu un arbre filogenètic sense arrel[5] per a la miosina, una família genètica de porteïnes.
Els dos tipus d'arbres filogenètics arrelats i sense arrels poden ser amb bifurcació o multifurcació, i també etiquetats o no etiquetats. Un arbre bifurcat té un màxim de dos descendents sortint de cada node interior, mentre que d'un multifurcat en sortirien més de dos. Un arbre etiquetat té assignats valors específics a les seves fulles, mentre que un no etiquetat només defineix una topologia. El nombre possible d'arbres per a un nombre donat de nòduls de fulles depèn del tipus d'arbre, però sempre hi ha més arbres multiforcats que biforcats, més etiquetats que no etiquetats, i més arrelats que sense arrels. La distinció final és la biològicament més rellevant; sorgeix perquè hi ha molts llocs en un arbre sense arrel per posar una arrel. Per arbres bifurcats etiquetats hi ha
total d'arbres arrelats i a
total d'arbres sense arrels, on n representa el nombre de nòduls de fulles. El nombre d'arbres sense arrel per a seqüències o espècies n és igual al nombre d'arbres arrelats per seqüències n-1.[6]
- Un Dendrograma és un terme genèric per a la representació diagràmatica d'un arbre filogenètic.
- Un Cladograma és un arbre format usant mètodes cladístics. Aquest tipus d'arbre només representa un patró de ramificació, és a dir, que la longitud de les seves branques no representen el temps.
- Un Filograma és un arbre filogenètic que representa explícitament un nombre de canvis de trets de caràcter al llarg de la longitud de les seves branques; és el resultat de l'aplicació dels principis de la sistemàtica evolutiva.
- Un Fenograma és un dendrograma no arrelat que s'estableixen les relacions de parentiu fenètic dels organismes estudiats; sorgeixen de l'aplicació dels mètodes de la taxonomia numèrica.
- Un Cronograma és un arbre filogenètic que representa explícitament el temps evolutiu al llarg de la longitud de les seves branques.
Construcció
[modifica]Els arbres filogenètics es construeixen usant diferents criteris: la distància, la parsimònia i la versemblança. Els mètodes basats en la distància, es tracta de triar el criteri de distància entre les futures fulles de l'arbre. Per exemple, si aquestes fulles són seqüències d'ADN, es pot triar com a distància entre dues fulles el nombre de nucleòtids en què difereixen. Per determinar aquest valor, s'ha d'efectuar un alineament de seqüències. Llavors es pot utilitzar el mètode UPGMA o el Neighbour Joining per deduir l'arbre.
Els mètodes de parsimònia consisteixen en trobar l'arbre que minimitza el nombre de mutacions, delecions, o insercions puntuals per passar d'una seqüència a una altra. Aquest mètode recerca, per tant, la xarxa més econòmica en substitucions. Així, si la llargada de les branques són proporcionals al nombre de substitucions sobrevingudes, se seleccionarà la xarxa que tingui la llargada total més curta. Aquest principi implica que els fenòmens d'evolució convergent i reversibilitat (retrocedir un caràcter a la condició ancestral) siguin relativament rars. D'aquesta manera, l'arbre que presenta un menor nombre de passes evolutives és el que minimitza l'existència d'aquests dos fenòmens.
Aquest mètode es divideix en tres etapes:
- A Recerca de tots els arbres filogenètics possibles per les diferents taxes estudiades,
- B mesutat la llargada total de cada arbre,
- C seleccionar aquell o aquells que presenten la llargada més petita.
Els arbres elaborats per aquest mètode són no polaritzats, no obstant la utilització de Out groups (espècie externa al grup estudiat) permet polaritzar l'arbre en un segon termini. És un mètode molt lent si es generen tots els arbre possibles per a calcular la parsimònia.
Finalment, els mètodes de versemblança són més probabilistes. Basant-se en la taxa de substitució per a cada element de base (nucleòtid per a les seqüències d'ADN) al llarg del temps, s'estima la versemblança de la posició i de la llargada de les branques de l'arbre.
Limitacions
[modifica]Encara que els arbres filogenètics produïts amb les dades de la seqüència genètica o genòmica de diferents espècies proporcionen sobre l'evolució, també tenen importants limitacions. No representen necessàriament amb exactitud la història evolutiva de les espècies, i de fet en la majoria dels casos no ho fan. Les dades en què es basen tenen soroll produïts per diversos factors com la transferència horitzontal de gens,[7] la hibridació entre les espècies que no eren veïnes més properes en l'arbre abans de la hibridació, l'evolució convergent, i conservació de seqüències de gens són elements de pertorbació que poden fàcilment confondre l'anàlisi basat en principis filogenètics.
Referències
[modifica]- ↑ Hodge T, Cope M «A myosin family tree». J Cell Sci, 113 Pt 19, 2000, pàg. 3353–4. PMID: 10984423.
- ↑ Letunic, I «Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation.» (PubMed). Bioinformatics, 23(1), 2007, pàg. 127–8.
- ↑ Ciccarelli, FD «Toward automatic reconstruction of a highly resolved tree of life.» (PubMed). Science, 311(5765), 2006, pàg. 1283–7. DOI: 10.1126/science.1123061. PMID: 16513982.
- ↑ Woese, C. R. 1998. The Universal Ancestor. Proceedings of the National Academy of Sciences 95: 6854-6859.
- ↑ Maher, B. A. 2002. Uprooting the Tree of Life. The Scientist 16: 18 (Sep. 16, 2002); subscription only Arxivat 2003-10-02 a Wayback Machine.
- ↑ Felsenstein J. (2004). Inferring Phylogenies Sinauer Associates: Sunderland, MA.
- ↑ Woese C «On the evolution of cells». Proc Natl Acad Sci U S A, 99, 13, 2002, pàg. 8742–7. DOI: 10.1073/pnas.132266999. PMID: 12077305.
Enllaços externs
[modifica]- Discover Life Arbre interactiu basat en el projecte de la U.S. National Science Foundation "Assembling the Tree of Life Project" (anglès)
- PhyloCode Arxivat 2007-11-14 a Wayback Machine. (anglès)
- Projecte Web de l'arbre de la vida (anglès)
- Arbre filogenètic dels dinosaures Arxivat 2011-09-12 a Wayback Machine. (anglès)
- Inferència filogenètica del T-REX server (anglès)
| Camps principals |  | |
|---|---|---|
| Conceptes bàsics | ||
| Mètodes d'inferència | ||
| Temes actuals | ||
| -morfia | ||
| -filetisme | ||